参考资料:Germline short variant discovery (SNPs + Indels)
下面这张图是单样本种系突变的主要分析步骤(Main steps for Germline Single-Sample Data)
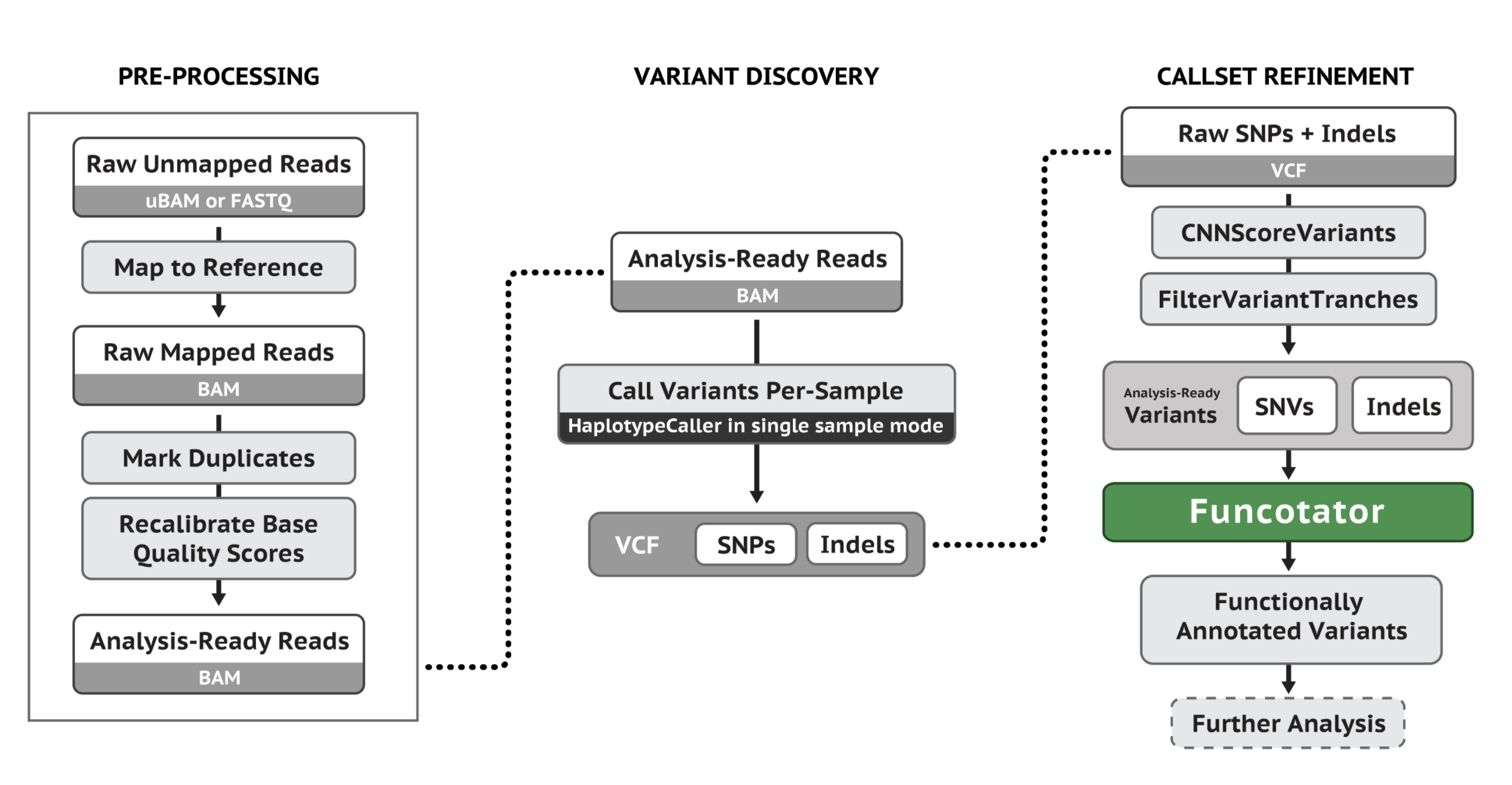
一、数据质控
1 | fastp -i /mnt/d/wes/sample/fastq/sam01_1.fastq -I /mnt/d/wes/sample/fastq/sam01_2.fastq -o /mnt/d/wes/sample/qc/clean_sam01_1.fastq -O /mnt/d/wes/sample/qc/clean_sam01_2.fastq |
二、比对至基因组
1 | bwa mem -t 4 -B 4 -R '@RG\tID:lane1\tSM:sample1\tLB:genome\tPL:ILLUMINA' -M ~/ncbi/hg38/chroms/hg38.fa /mnt/d/wes/sample/qc/clean_sam01_1.fastq /mnt/d/wes/sample/qc/clean_sam01_2.fastq | samtools view -q4 -bS -o - - | samtools sort -@4 -o /mnt/d/wes/sample/bam/clean_sam01.bam - |
三、PCR去重
1 | gatk MarkDuplicates -I /mnt/d/wes/sample/bam/clean_sam01.bam -O /mnt/d/wes/sample/nodup/nodup_sam01.bam -M /mnt/d/wes/sample/nodup/metrics_sam01.txt --REMOVE_DUPLICATES true --ASSUME_SORTED true |
四、BQSR
1 | # 第一步 |
五、变异检测
1 | gatk HaplotypeCaller -R /home/eric/ncbi/hg38/chroms/hg38.fa -I /mnt/d/wes/sample/bqsr/bqsr_sam01.bam -O /mnt/d/wes/sample/caller/output.g.vcf.gz -ERC GVCF |
六、位点注释
位点注释需要用到三个工具:CNNScoreVariants和FilterVariantTranches,以及Funcotator
CNNScoreVariants是指使用CNN (Convolutional Neural Network)的分数对VCF文件进行注释,这个工具需要调用PYTHON程序,因此,需要预先搭建环境,最简单不易出错的方法是使用Docker,关于Docker的搭建及测试会专门写一篇文章介绍。
6.1 启动Docker
1 | # 由于在Docker内部无法调用外部文件,因此,需要将外部文件挂载在Docker中,如果把Docker看作是一个独立的操作系统,挂载后的外部文件即相当于Docker内部的一个盘符,方便调用,在此,使用-V参数将D盘挂载到Docker下,并命名为my_data目录,路径为"/gatk/my_data/";同时,使用-it参数启动gatk。 |
6.2 使用CNNScoreVariants进行评分
1 | gatk CNNScoreVariants -V /gatk/my_data/wes/sample/caller/output.vcf.gz -R /gatk/my_data/ncbi/hg38/chroms/hg38.fa -O /gatk/my_data/wes/sample/annotate/annotated.vcf --disable-avx-check true |
6.3 使用FilterVariantTranches过滤
1 | # 这个工具根据注释后的VCF中的INFO字段的分数进行过滤,注释可以是CNNScoreVariants(CNNLOD)或VQSR (VQSLOD) |
6.4 使用Funcotator进行功能注释
在使用Funcotator进行功能注释前,需要下载Data Sources。GATK有两套预封装数据源,分别对应germline和somatic,可以使用gatk FuncotatorDataSourceDownloader工具进行下载
1 | # For somatic data sources: (29GB) |
数据源下载完成后即可进行分析,语法示例:
1 | ./gatk Funcotator \ |
在本例中,使用如下命令:
1 | # Germline Mutation |
至此,我们得到了注释好的VCF文件,下面,就可以进行后续的功能分析了。
