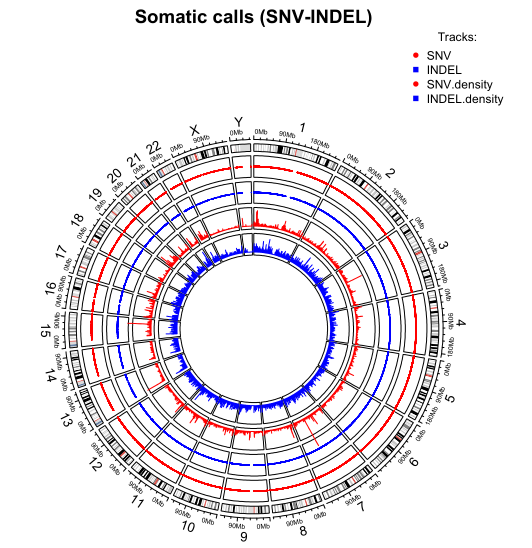
R package circlize:可惜这个文档写得太枯燥。下面作图主要参考这个教程Variant Visualisation
我们的目的是使用CIRCLIZE这个程序来对VCF文件作图,即对VCF进行简单的可视化,显示SNP或INDEL在染色体上的位置。
1 | > library(circlize) |
上面在作图时,实际上我们只标示了在染色体何位置上有SNP或INDEL,但不能显示差异,我们还可以对SNP或INDEL的密度作图,计算密度可以使用VCFTOOLS,
1 | > vcftools --vcf snp.vcf --SNPdensity 100000 --out snp |
回到R中,
1 | > snpden <- read.table("./snp.snpden",sep="\t",header=T) |
对于INDEL,也是如此,
1 | > indelden <- read.table("./indel.snpden",sep="\t",header=T) |