关于变异检测,目前有多个程序可供使用,但很难说哪个程序更好,需要用实验的手段来验证。目前,TCGA采取4款软件,varscan,MuTect,MuSE,SomaticSniper。在这个教程中,我们会陆续介绍这几款主流的程序的使用方法。
一、Varscan
1 | $ samtools mpileup -d 1000 -B -q 1 -f /mnt/d/ncbi/hg19/BWAIndex/hg19.fa /mnt/d/cancer/bqsr/SRR5478492C.bqsr.nodup.bam /mnt/d/cancer/bqsr/SRR5478491C.bqsr.nodup.bam > /mnt/d/cancer/variant/normal-tumor.mpileup |
现在,可以使用进行VARIANT CALLING
varscan是基于启发式的算法,并通过对一组样本进行统计检验来检测变异,可用于检测somatic/germline variants或CNV
1 | ***NON-COMMERCIAL VERSION*** |
对于somatic/germline variants,可以使用varscan somatic命令
1 | USAGE: VarScan somatic [normal_pileup] [tumor_pileup] [Opt: output] OPTIONS |
二、MuTect2
MuTect2使用起来非常繁琐,软件更新十分频繁,因此流程变来变去,不推荐使用。详情敬请移步官方教程:
Call somatic mutations using GATK4 Mutect2;
Mutect2;
Somatic short variant discovery (SNVs + Indels)
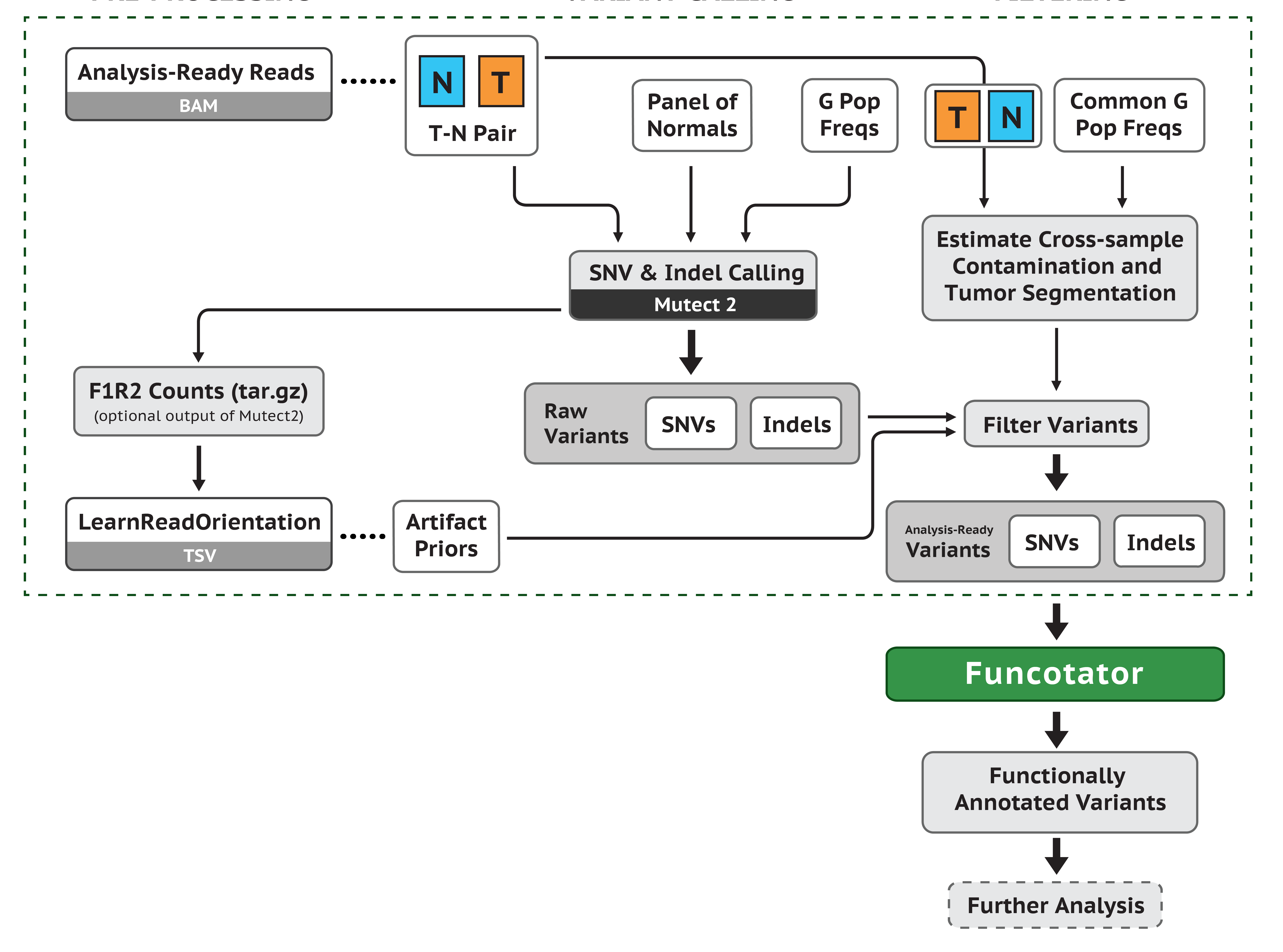
2.1 创建PON (panel of normals)
1 | $ gatk Mutect2 -R /mnt/d/ncbi/hg19/BWAIndex/hg19.fa -I /mnt/d/cancer/bqsr/SRR5478492C.bqsr.nodup.bam --max-mnp-distance 0 -O /mnt/d/cancer/variant/mutect/normal.vcf.gz |
第三步中,需要用到一个af-only-gnomad.hg19.vcf.gz文件,如果使用hg38参考基因组,这个文件可以从gatk的resource of bundle上下载,但是对于hg19,并不提供这个文件。但是,我们可以找到gnomAD的完全版,8.3G,我们可以使用bcftools来提取AF信息,并生成新的文件,过程如下:
下载地址: hg19
1 | # 下载后的文件 |
2.2 获取原始的候选变异
1 | gatk Mutect2 -R /mnt/d/ncbi/hg19/BWAIndex/hg19.fa -I /mnt/d/cancer/bqsr/SRR5478491C.bqsr.nodup.bam -I /mnt/d/cancer/bqsr/SRR5478492C.bqsr.nodup.bam -normal SRR5478492C.bqsr --germline-resource af-only-gnomad.vcf.gz --panel-of-normals pon.vcf.gz -O somatic.vcf.gz |
https://hgdownload.soe.ucsc.edu/gbdb/hg19/gnomAD/vcf/
https://console.cloud.google.com/storage/browser/gatk-best-practices/somatic-b37;tab=objects?pageState=(%22StorageObjectListTable%22:(%22f%22:%22%255B%255D%22))&prefix=&forceOnObjectsSortingFiltering=false
https://gatk.broadinstitute.org/hc/en-us/articles/360035890711-GRCh37-hg19-b37-humanG1Kv37-Human-Reference-Discrepancies
https://gatk.broadinstitute.org/hc/en-us/articles/360035890951-Human-genome-reference-builds-GRCh38-or-hg38-b37-hg19
https://gatk.broadinstitute.org/hc/en-us/articles/360035894731-Somatic-short-variant-discovery-SNVs-Indels-
https://blog.csdn.net/viancheng/article/details/106332585
拷贝数变异
生成GC content文件
1 | $ sequenza-utils gc_wiggle --fasta /mnt/d/ncbi/hg19/BWAIndex/hg19.fa -w 50 -o /mnt/d/ncbi/wxs/sequenza/hg19_gc50.wig.gz |
sequenza-utils bam2seqz –normal normal_sample.bam –tumor tumor_sample.bam \
–fasta genome.fa.gz -gc genome_gc50.wig.gz –output sample.seqz.gz
sequenza bam2seqz \
-n data/normal.chr5.60Mb.bam \
-t data/tumour.chr5.60Mb.bam \
–fasta assets/human_g1k_v37.fasta \
-gc assets/human_g1k_v37.gc50Base.txt.gz \
-C 5:1-60000000 | gzip > stage1.seqz.gz
-n: the normal BAM
-t: the tumour BAM
–fasta: the reference genome used for mapping (b37 here)
-gc: GC content as windows through the genome (pre-generated and downloadable from the Sequenza website)
-C: specifies the genomic location to process
$ sequenza-utils bam2seqz –normal /mnt/d/cancer/bqsr/SRR5478492C.bqsr.nodup.bam -t /mnt/d/cancer/bqsr/SRR5478491C.bqsr.nodup.bam –fasta /mnt/d/ncbi/hg19/BWAIndex/hg19.fa -gc /mnt/d/ncbi/wxs/sequenza/hg19_gc50.wig.gz –output /mnt/d/cancer/cnv/stage1.seqz.gz
$ sequenza-utils bam2seqz –normal /mnt/d/cancer/bqsr/SRR5478492C.bqsr.nodup.bam -t /mnt/d/cancer/bqsr/SRR5478491C.bqsr.nodup.bam –fasta /mnt/d/ncbi/hg19/BWAIndex/hg19.fa -gc /mnt/d/ncbi/wxs/sequenza/hg19_gc50.wig.gz –output /mnt/d/cancer/cnv/stage1.seqz.gz
[mpileup] 1 samples in 1 input files
[mpileup] 1 samples in 1 input files
total 75M
-rwxrwxrwx 1 eric eric 75M Nov 27 22:47 stage1.seqz.gz
-rwxrwxrwx 1 eric eric 126K Nov 27 22:47 stage1.seqz.gz.tbi
$ zcat stage1.seqz.gz | head -n 20
chromosome position base.ref depth.normal depth.tumor depth.ratio Af Bf zygosity.normal GC.percent good.reads AB.normal AB.tumor tumor.strand
chr1 69457 C 13 7 0.538 1.0 0 hom 48 7 C . 0
chr1 69458 T 13 7 0.538 1.0 0 hom 48 7 T . 0
chr1 69459 A 13 7 0.538 1.0 0 hom 48 7 A . 0
chr1 69460 C 14 8 0.571 1.0 0 hom 48 8 C . 0
chr1 69461 A 14 8 0.571 1.0 0 hom 48 8 A . 0
chr1 69462 C 14 8 0.571 1.0 0 hom 48 8 C . 0
chr1 69463 T 14 8 0.571 1.0 0 hom 48 8 T . 0
chr1 69464 A 15 8 0.533 1.0 0 hom 48 8 A . 0
chr1 69465 C 16 8 0.5 1.0 0 hom 48 8 C . 0
chr1 69466 A 17 9 0.529 1.0 0 hom 48 9 A . 0
chr1 69467 C 17 9 0.529 1.0 0 hom 48 9 C . 0
chr1 69468 T 17 9 0.529 1.0 0 hom 48 9 T . 0
chr1 69469 A 16 9 0.562 1.0 0 hom 48 9 A . 0
chr1 69470 C 18 9 0.5 1.0 0 hom 48 9 C . 0
chr1 69471 A 20 9 0.45 1.0 0 hom 48 9 A . 0
chr1 69472 A 22 9 0.409 1.0 0 hom 48 9 A . 0
chr1 69473 T 22 11 0.5 1.0 0 hom 48 11 T . 0
chr1 69474 T 22 14 0.636 1.0 0 hom 48 14 T . 0
chr1 69475 A 22 14 0.636 1.0 0 hom 48 14 A . 0
sequenza-utils seqz-binning \
-w 200 \
-s stage1.seqz.gz | gzip > stage2.seqz.gz
sequenza-utils seqz_binning -w 200 -s stage1.seqz.gz -o stage2.seqz.gz
1 | > library(sequenza) |
