本教程目的是对上一步生成的BAM文件进行质控,并检测变异,以及对变异进行注释,参考教程:Single Nucleotide Variant Calling and Annotation,Data pre-processing for variant discovery,以及Somatic short variant discovery (SNVs + Indels)
一、查看BAM信息
我们先来具体看一下BAM文件,下面是BAM文件主体内容的前4行
1 | $ samtools view SRR5478491C.bam | head -n 4 |
SAM/BAM文件至少有11列,内容如下
1 | # 1. QNAME, Query template NAME |
特别关注一下第6列,此列名为CIGAR,比如,13M1I87M,代表如下信息:匹配13个碱基,第14个碱基是插入,从15至101(87个)碱基匹配,当然,M可以表示匹配也可表示SNP,即SNP也会被标记为M
1 | M == base aligns but doesn’t have to be a match. A SNP will have an M even if it disagrees with the reference. |
关于SAM//BAM文件的格式,可参见其 说明文件或SAM;关于FLAG,可参看FLAG,通过查询,可知上面的163代表:
1 | read paired (0x1) |
查看不配对的reads,可以使用samtools view -c -f4实现,若查看配对的reads,可以使用samtools view -c -F4实现
1 | # -c print only the count of matching records # 打印数目 |
二、BAM去除DUPLICATES
现在,我们得到的BAM文件称为RAW BAM,需要进行预处理,主要包括去掉PCR重复和BQSR,如下图
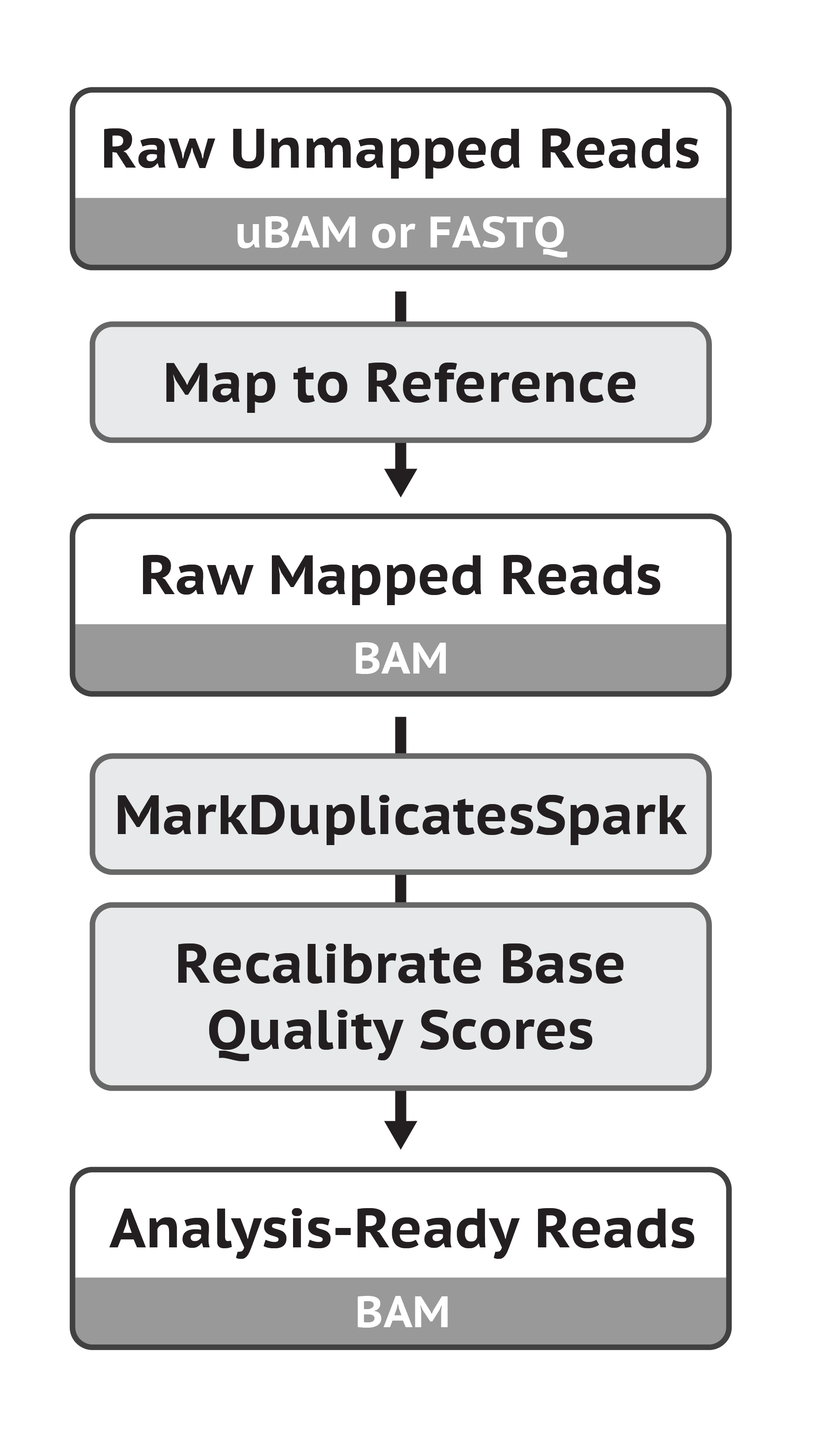
PCR去重可以通过MarkDuplicates来实现,即使用MarkDuplicates来标记重复,并将其直接去掉,不输出到BAM文件中,再使用samtools index生成索引文件。如果电脑性能比较强悍,建议使用最新的MarkDuplicatesSpark
1 | $ gatk MarkDuplicates -I /mnt/d/cancer/bam/SRR5478491C.bam -O /mnt/d/cancer/nodup/SRR5478491C.nodup.bam -M /mnt/d/cancer/nodup/SRR5478491C.metrics.txt --REMOVE_DUPLICATES true --ASSUME_SORTED true --CREATE_INDEX true |
三、校正碱基质量
现在,我们得到的是去掉PCR重复的BAM文件,下面进行Base (Quality Score) Recalibration (BQSR),即碱基质量校正,原因在于,测序过程中不同厂商的碱基读取有不同的倾向,需要重新校正,此步是通过机器学习来对测序结果中的碱基质量进行校正,通过两步实现:
- Build covariates based on context and known SNP sites. 根据物种已知SNP,生成用于质量校准的metrics
- Correct the reads based on these metrics. 根据metrics文件生成新的BAM文件
首先,需要先为hg19.fa生成dict文件,这可以通过调用CreateSequenceDictonary模块来实现
1 | $ gatk CreateSequenceDictionary -R hg19.fa -O hg19.dict |
3.1 BQSR step1,BaseRecalibrator
1 | $ gatk BaseRecalibrator -I /mnt/d/cancer/nodup/SRR5478491C.nodup.bam -R /mnt/d/ncbi/hg19/BWAIndex/hg19.fa --known-sites /mnt/d/ncbi/hg19/bqsr/1000G_phase1.snps.high_confidence.hg19.sites.vcf.gz --known-sites /mnt/d/ncbi/hg19/bqsr/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf.gz --known-sites /mnt/d/ncbi/hg19/bqsr/dbsnp_138.hg19.vcf.gz -L /mnt/d/ncbi/wxs/agilent/sureSelect_All_Exon_V5/S04380110_Padded.bed -O /mnt/d/cancer/bqsr/recal_SRR5478491C.table |
–known-sites中用到的三个VCF文件,用于提供物种已知的多态性位点,这些位点将被分析程序跳过,下载地址resources bundle of hg19,下载好的文件可以解压后使用,但解压文件空间占用太大,建议重新压缩并生成索引后使用
1 | $ bgzip -d ***.vcf.gz |
3.2 BQSR step2,ApplyBQSR
1 | $ gatk ApplyBQSR -I /mnt/d/cancer/nodup/SRR5478491C.nodup.bam -R /mnt/d/ncbi/hg19/BWAIndex/hg19.fa --bqsr-recal-file /mnt/d/cancer/bqsr/recal_SRR5478491C.table -L /mnt/d/ncbi/wxs/agilent/sureSelect_All_Exon_V5/S04380110_Padded.bed -O /mnt/d/cancer/bqsr/SRR5478491C.bqsr.nodup.bam --create-output-bam-index true |
四、BAM文件质控
4.1 计算覆盖度
1 | # 使用[DepthOfCoverage](https://gatk.broadinstitute.org/hc/en-us/articles/360046789152-DepthOfCoverage-BETA-)可以计算BAM文件的覆盖度 |
运行后看一下覆盖度
1 | $ less SRR5478491C.bqsr.nodup.coverage.sample_summary |
4.2 计算Insert Size
1 | gatk CollectInsertSizeMetrics \ |
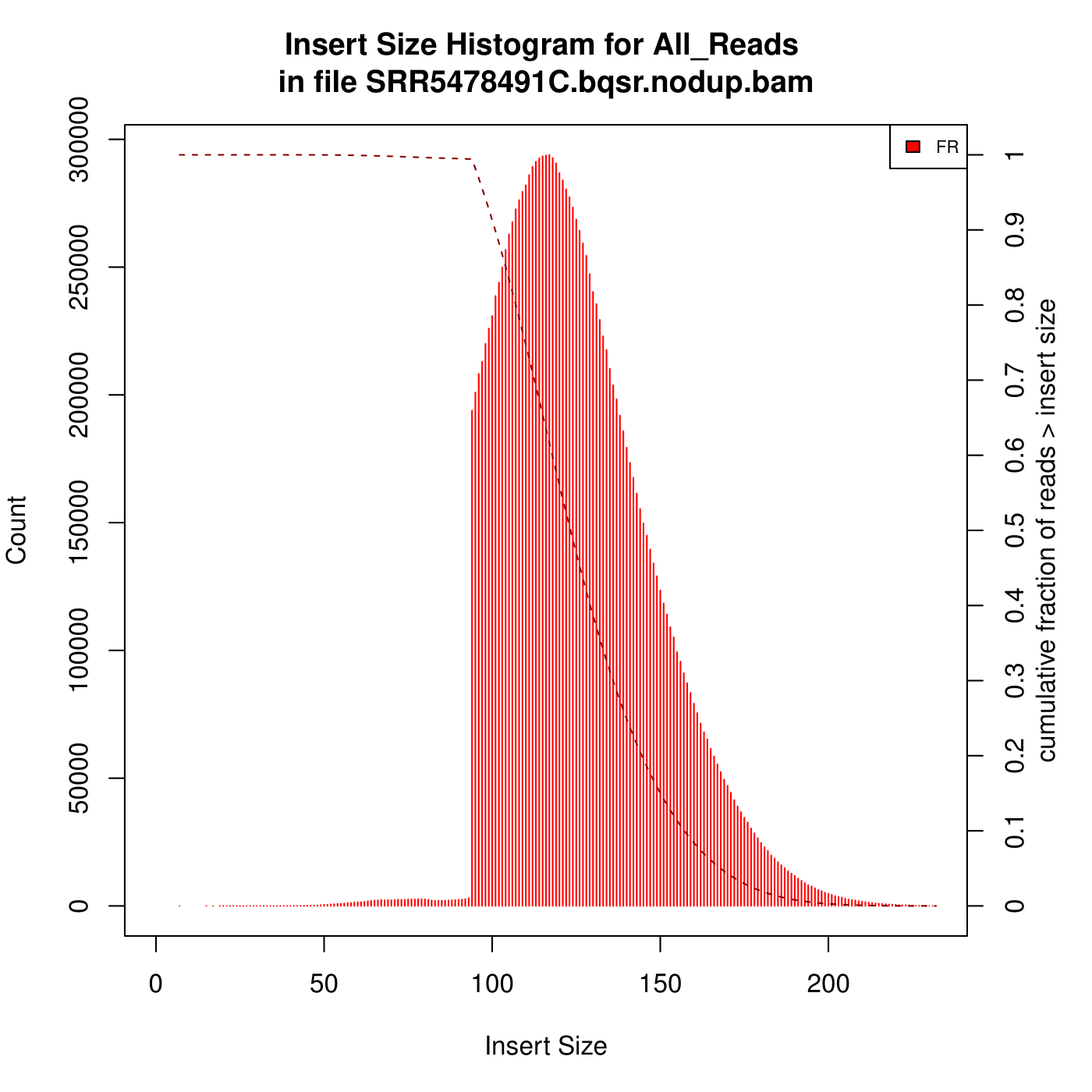
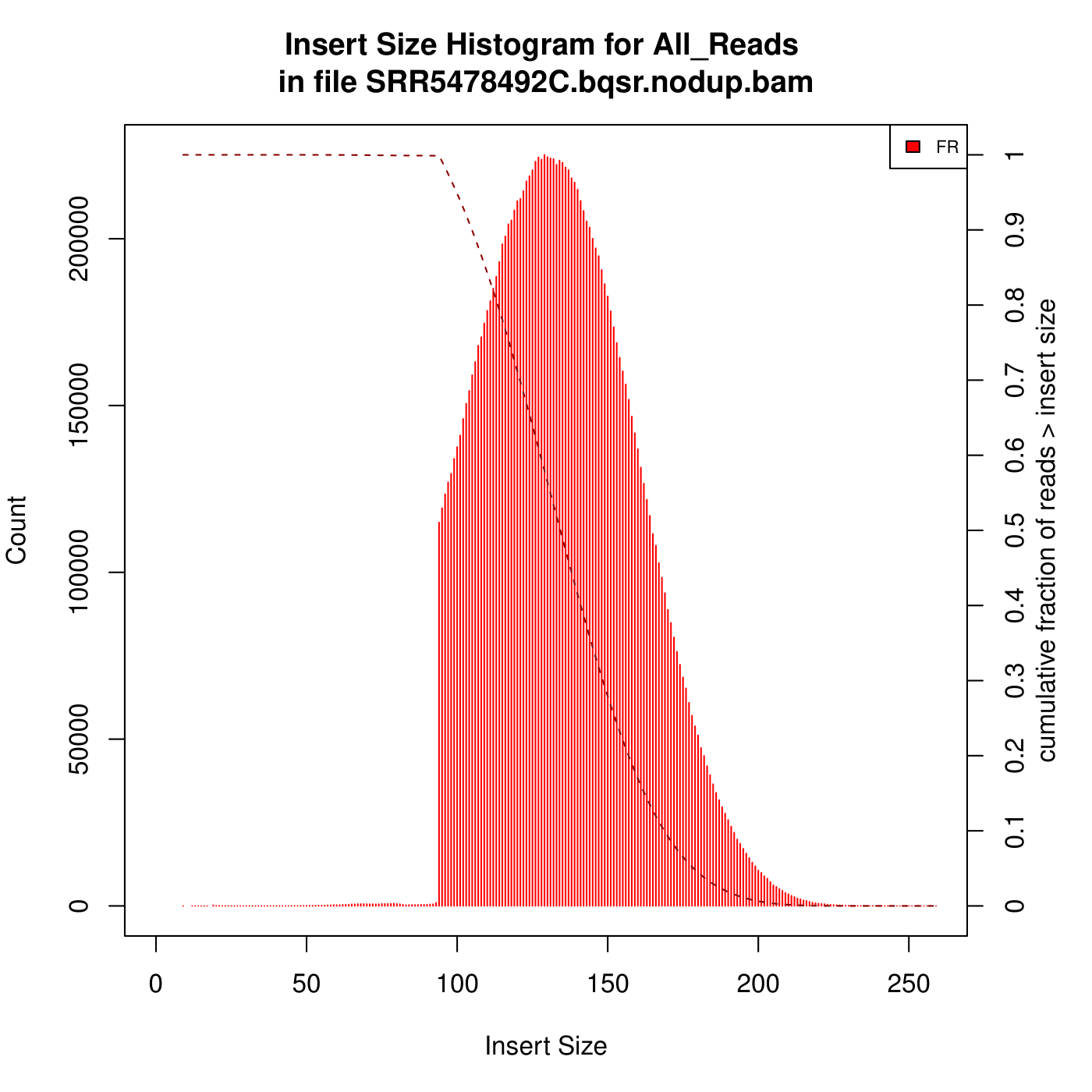
4.3 计算Alignment metrics
1 | gatk CollectAlignmentSummaryMetrics \ |
关于BAM文件的处理与质控就到此为止,我们现在得到了“Analysis-Ready Reads”的BAM文件,下一步就可以进行MUTATION CALLING。
