本教程主要目的是下载测序数据,并将其比对至基因组上,参考教程:Read Alignment。
主要流程见下图
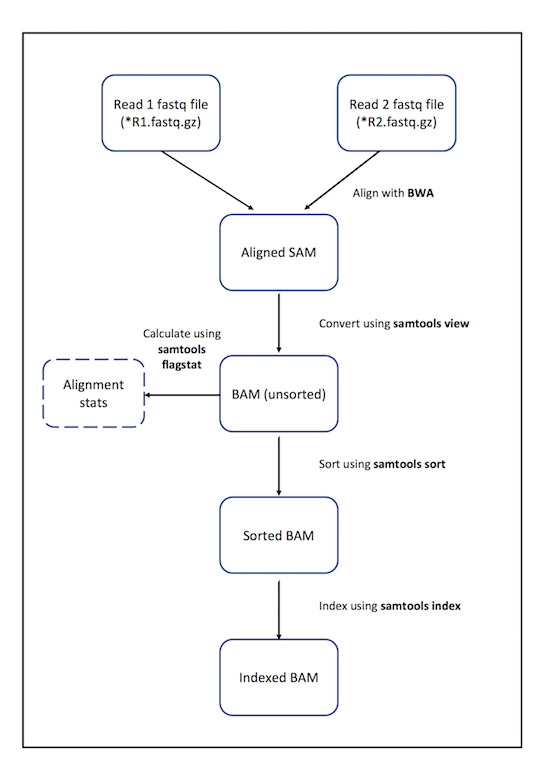
一、下载数据
本次分析使用的数据来自SRA数据库,患有ccRCC的病人,分析其中的两个样本,SRR5478491 (matched primary ccRCC)与 SRR5478492 (matched normal),双端测序,WXS,平台是ILLUMINA,Library Name是P1T与P1N,参考基因组为GRCh37。
1 | # 下载数据 |
二、FASTQ质控
获得FASTQ文件之后,按老规矩做个质控
1 | $ fastqc -f fastq SRR5478491_1.fastq.gz |
记录一下文件大小,后面对比一下
1 | $ ls -lh |
来看一下FASTQ文件的质量,
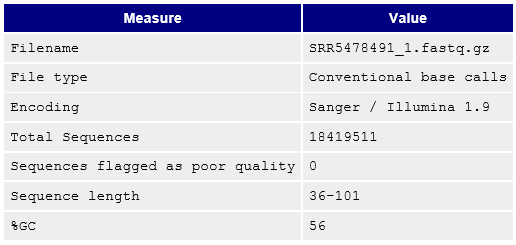
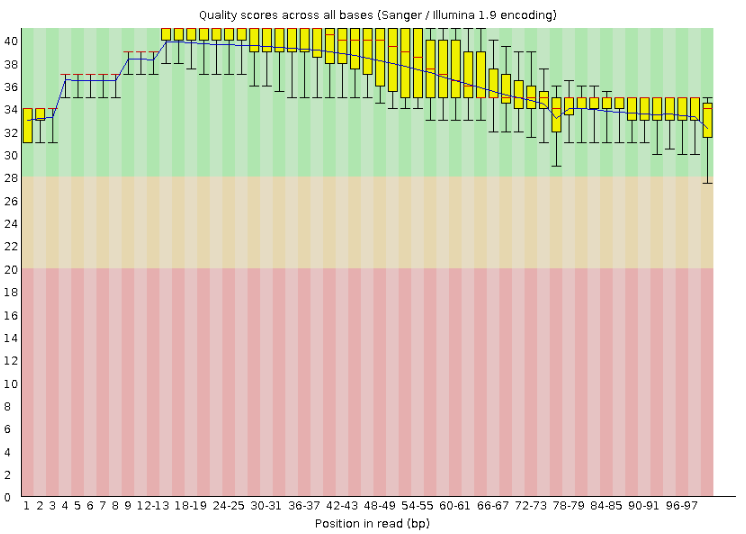
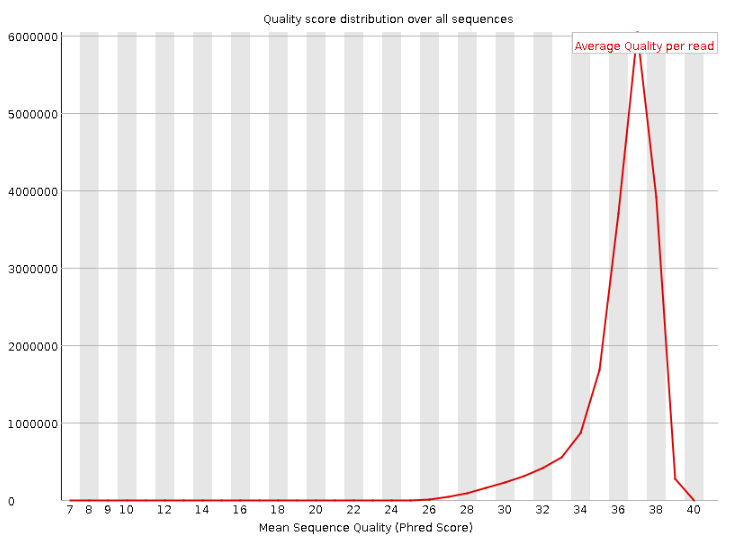
根据统计结果,测序数据质量还是不错的,长度在36-101之间,质量也很不错,而且没有接头,可见已经做过质控,这里作为演示,我们使用fastp做个质控。
1 | $ fastp -z 4 -i ./SRR5478491_1.fastq.gz -I ./SRR5478491_2.fastq.gz -o ./qc/SRR5478491C_1.fastq.gz -O ./qc/SRR5478491C_2.fastq.gz |
质控后文件变大一点
1 | -rwxrwxrwx 1 eric eric 1.1G Nov 21 20:25 SRR5478491C_1.fastq.gz |
二、使用BWA比对至基因组
BWA适合将低差异度的序列比对到大型基因组上,有三种算法: BWA-backtrack, BWA-SW 和 BWA-MEM。第一种算法用于比对100bp以内的ILLUMINA序列,后两种算法可以比对更长的序列(70bp-1Mbp),后两者相比,BWA-MEM更新、更快、更准确。而且,即使对于70bp-100bp的序列,BWA-MEM相比BWA-backtrack也有更好的表现。
BWA使用索引基因组来进行比对,以占用更小的内存,对于特定的基因组版本,如需产生索引,可以使用如下命令:
1 | $ bwa index hg19.fa |
生成索引文件之后,我们开始使用BWA进行比对
1 | $ mkdir bam # 创建存放BAM文件的目录 |
比对生成SAM文件,但是SAM文件非常大,太耗空间,因此,将其转化为等价的二进制BAM文件,使用命令samtools view实现
1 | # -q,线程数 |
此步最耗时间,运行记录如下:
1 | $ bwa mem -M -t 4 -R '@RG\tSM:ccRCC\tID:SRR5478491\tLB:P1T\tPL:ILLUMINA' /mnt/d/ncbi/hg19/BWAIndex/hg19.fa /mnt/d/cancer/qc/SRR5478491C_1.fastq.gz /mnt/d/cancer/qc/SRR5478491C_2.fastq.gz | samtools view -q4 -bS -o - - | samtools sort -@2 -o /mnt/d/cancer/bam/SRR5478491C.bam - |
生成的BAM文件需要索引,而建立索引则需要对BAM文件按染色体进行排序,可以使用samtools view -h来查看BAM文件头部,确定是否已经排序
1 | $ samtools view -h SRR5478491C.bam | less |
看一下文件大小
1 | total 4.1G |
使用samtools flagstat来对Flag values做个统计,每一项统计数据都由两部分组成,即QC-passed与QC-failed,每一项都有详细说明,不重复列举
1 | $ samtools flagstat SRR5478491C.bam |
