本教程目的是对数据质控有初步理解,并熟悉以下工具的使用:FastQC,Skewer,FASTX-Toolkit。参考教程:Quality Control
在高通量测序过程中,很难避免引入测序错误,如碱基读取错误(base calling errors)、插入或缺失(small insertions/deletions),低质量碱基(poor quality reads)、引物与接头污染(primer/adapter contamination),以及最常见的碱基替换(substitution),错误发生频率约 0.5-2.0%,且多发生于3’区域。
因此,在数据分析之前,有必要对原始数据进行质量控制,如去除低质量碱基(trimming off low quality bases)、去除接头(cleaning up any sequencing adapters)、去除PCR重复(removing PCR duplicates),另外,在深度测序(> 15X)时,部分测序错误可以通过算法被修正。
一、质量值编码方案(Quality Value Encoding Schema)
质量值是测序中非常重要的参考标准,使用Phred编码方案,质量编码体现在FASTQ格式的第四行,如
1 | @HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 |
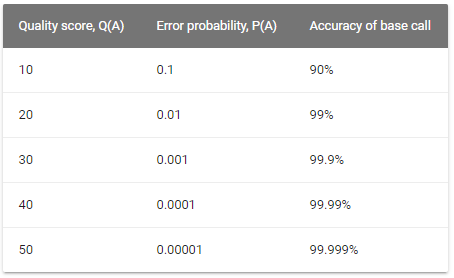
碱基质量可以通过查表得出对应的Q-score,Q(A)=−10log10(P(A)),A表示错误概率,如下表所示:
二、FASTQ
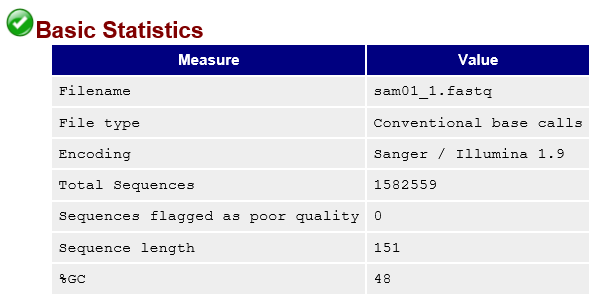
现在我们的目录下有两个fastq文件,sam01_1.fastq & sam01_2.fastq,其实是来自同一个样本,双端测序生成的两个文件,我们只对其中一个检查质量
1 | $ fastqc -f fastq sam01_1.fastq |
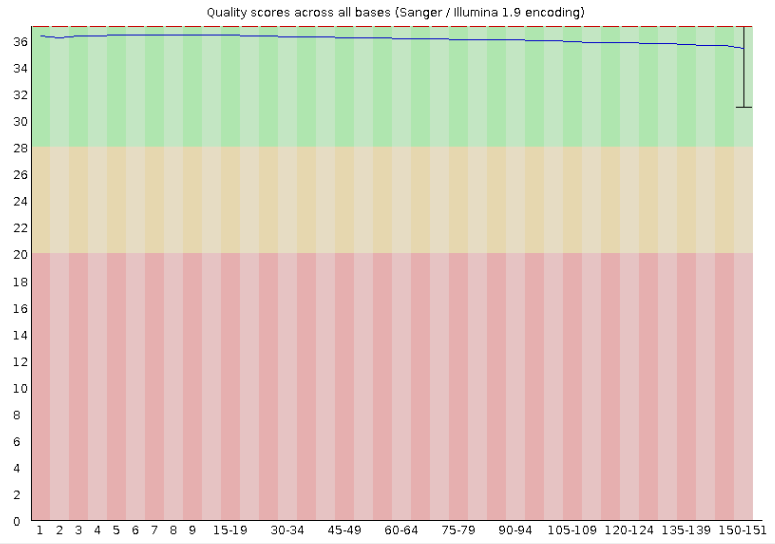
每个碱基的平均质量Q-score,也即前面提到的Phred值,这个测序结果质量相当好,平均质量在34以上,只有尾部稍差,也有30以上。
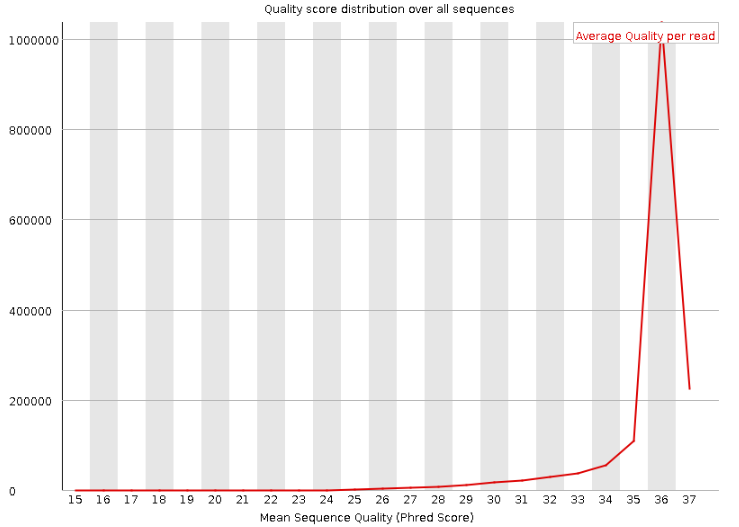
序列对应的质量分数,可以看到,绝大多数序列的Q值都很高,平均值约36
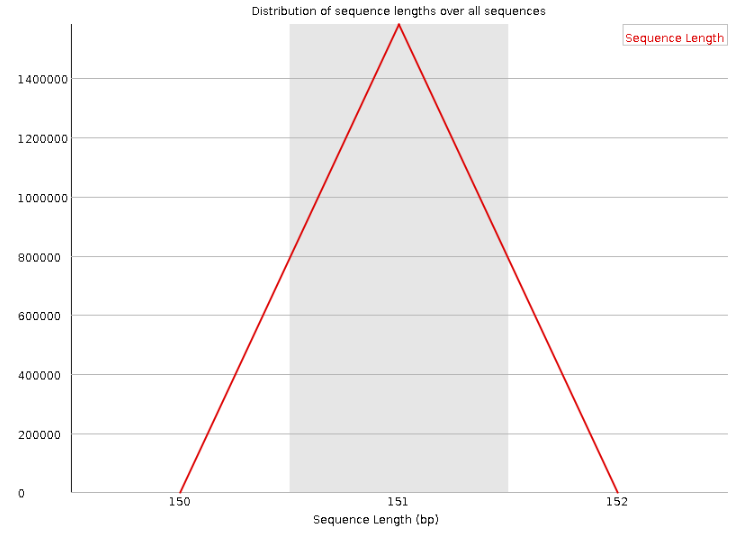
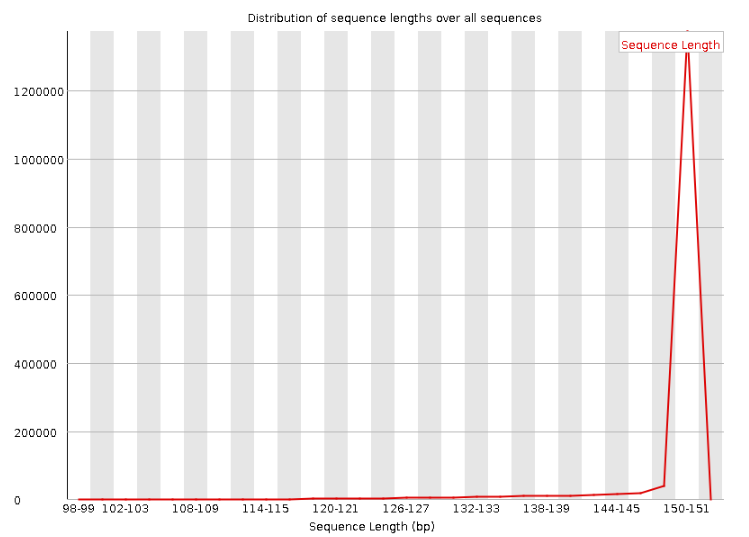
如果测序质量不好,则可以通过Read Trimming来去除低质量的读段。下面这张图显示的是读长,基本上是151 bp
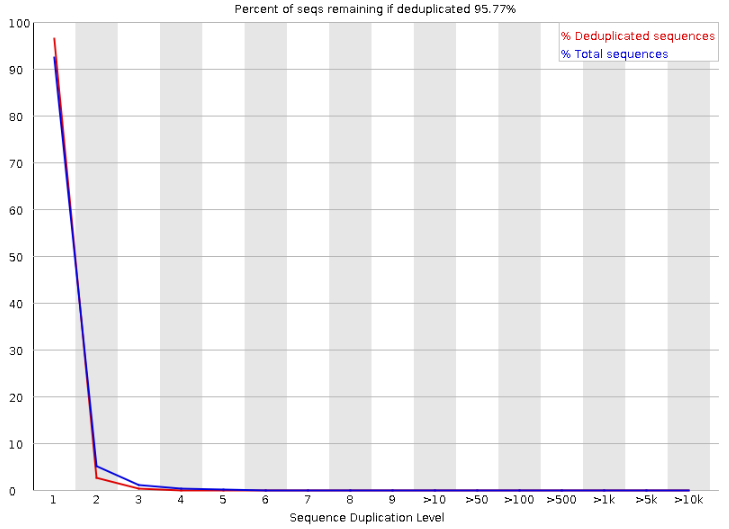
可以看到有重复,后面会去掉
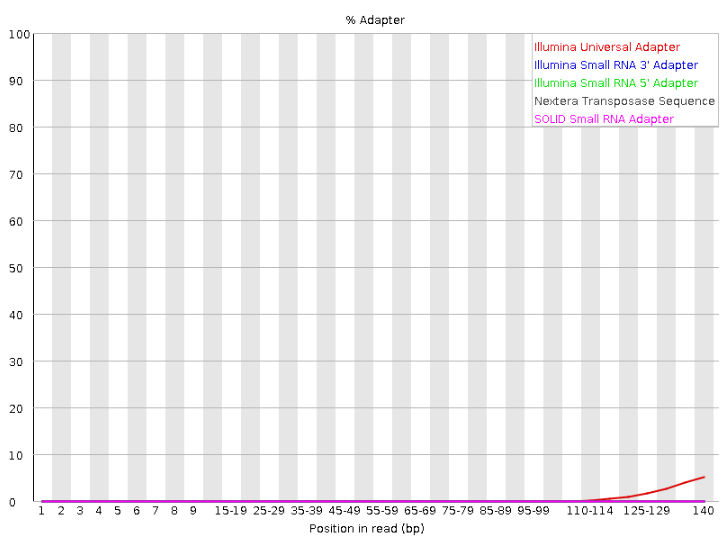
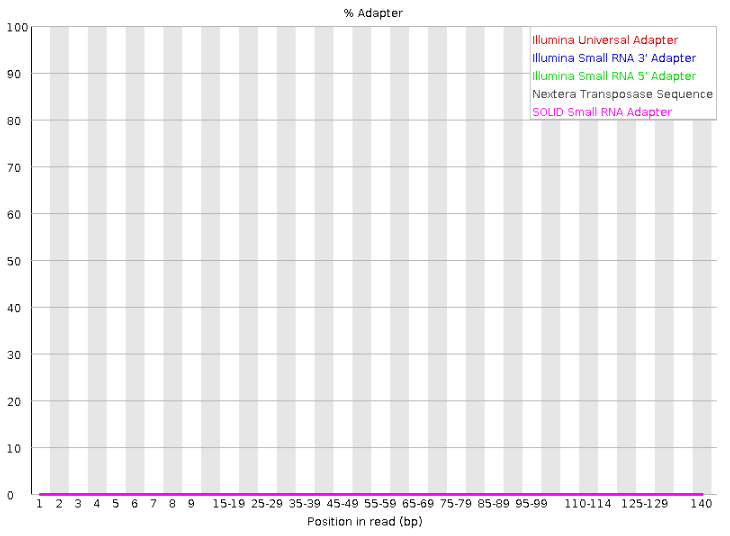
3’端有ILLUMINA的通用接头,红色的线
三、Read Trimming
1 | $ conda install -c bioconda skewer # 安装skewer |
来看一下发生哪些变化,
1 | -rwxrwxrwx 1 eric eric 525M Nov 3 11:53 sam01_1.fastq |
对TRIM后的文件查看质量
1 | $ fastqc -f fastq trim-trimmed-pair1.fastq |
观察一下,发生什么变化?首先,统计信息改变,总长度变为1563563(98.8%),跟skewer的日志中提供的信息一致,序列长度由151变为100-151,GC含量不变。
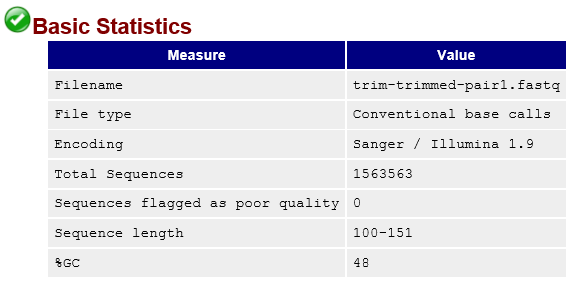
3’端的低质量碱基被过滤除掉
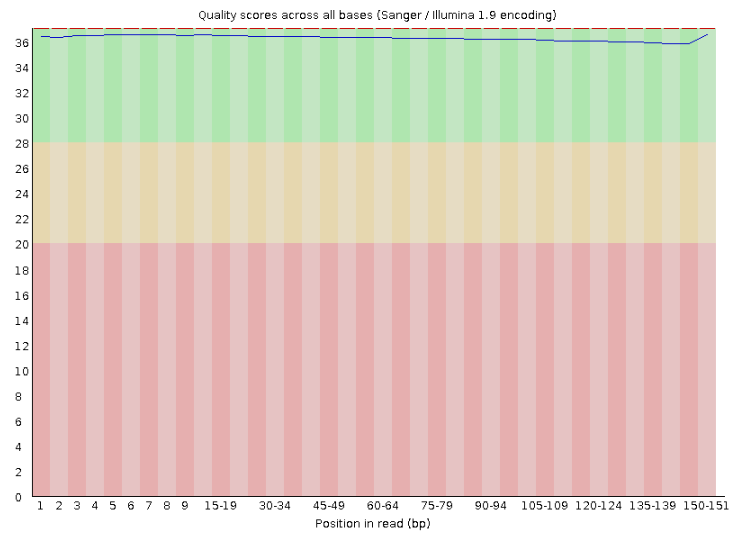
读长变为100-151
重复还在,可见不是这步被除掉的
接头序列被过滤除掉
假如,在建库过程中使用了特定的adaptor,你知道序列,但是没有被程序自动过滤除掉,则可以使用skewer来去掉接头,如接头序列是TGGAATTCTCGGGTGCCAAGGT,
1 | skewer -x TGGAATTCTCGGGTGCCAAGGT -t 20 -l 10 -L 35 -q 30 adaptorQC.fastq.gz |
另外,也可以锁定长度区间来FASTQ文件进行TRIM,这里不作介绍。
