上面,我们根据差异表达提取出来的4913个基因在611个样品中的表达数据进行了主成份分析,根据主成份成分的结果,正常与肿瘤样本能很好地分成两类,这对样本的预测是很好的辅助。下面,我们将根据差异表达的结果来绘制热图,看能否找出预示肿瘤组织的标志分子。
1 | > library(gplots) # 在R中,绘制热图的程序有很多,这里我们使用gplots::heatmap.2; |
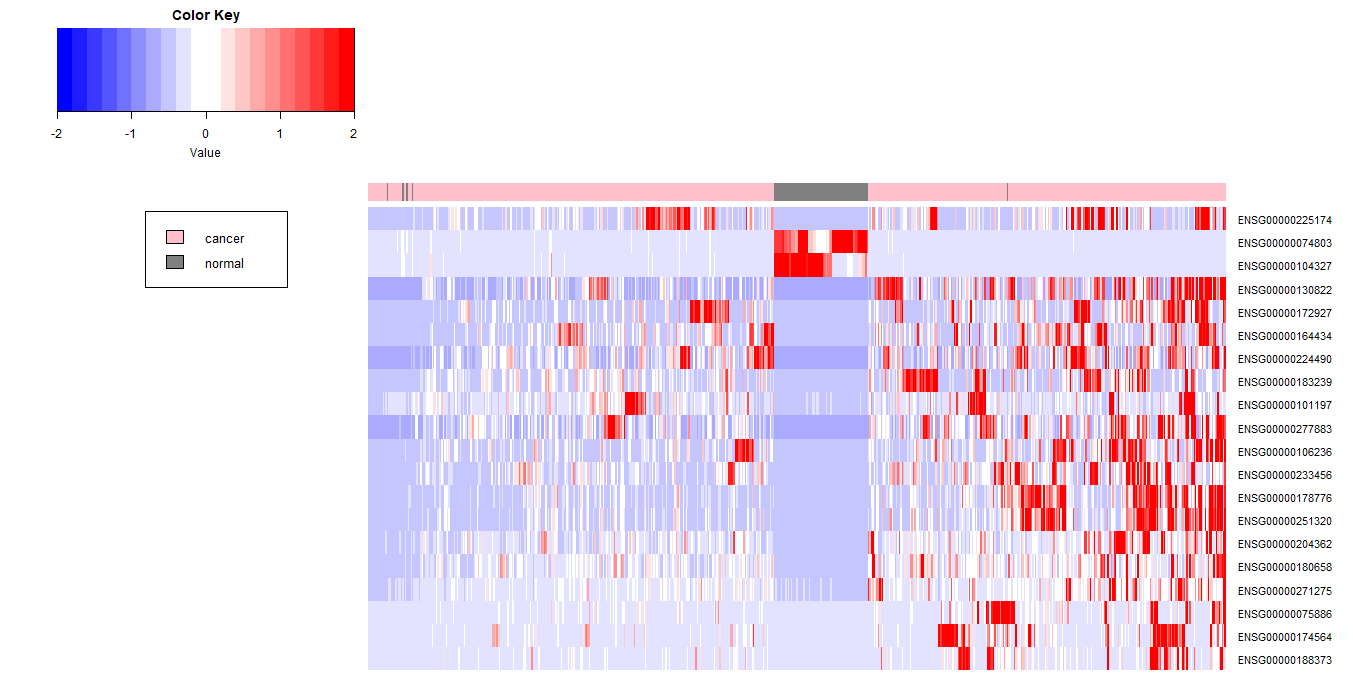
下面,我们使用TOP50再来绘制热图
1 | > result_select_top50 <- result_select_FCOrder[1:50,] |
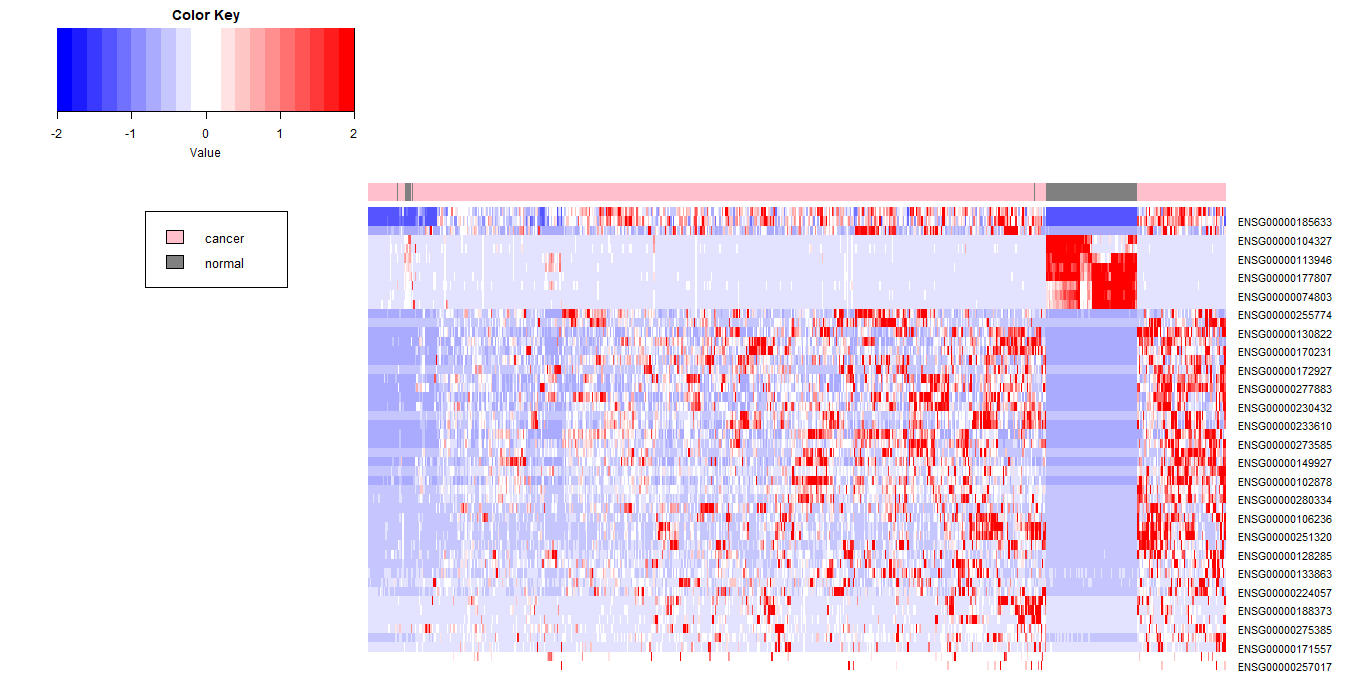
如果将标准化后的值限定在3以内,看一下图像的变化,颜色会变淡一些
1 | heat_scaled_limit3 <- ifelse(heat_scaled>3,3,heat_scaled) |
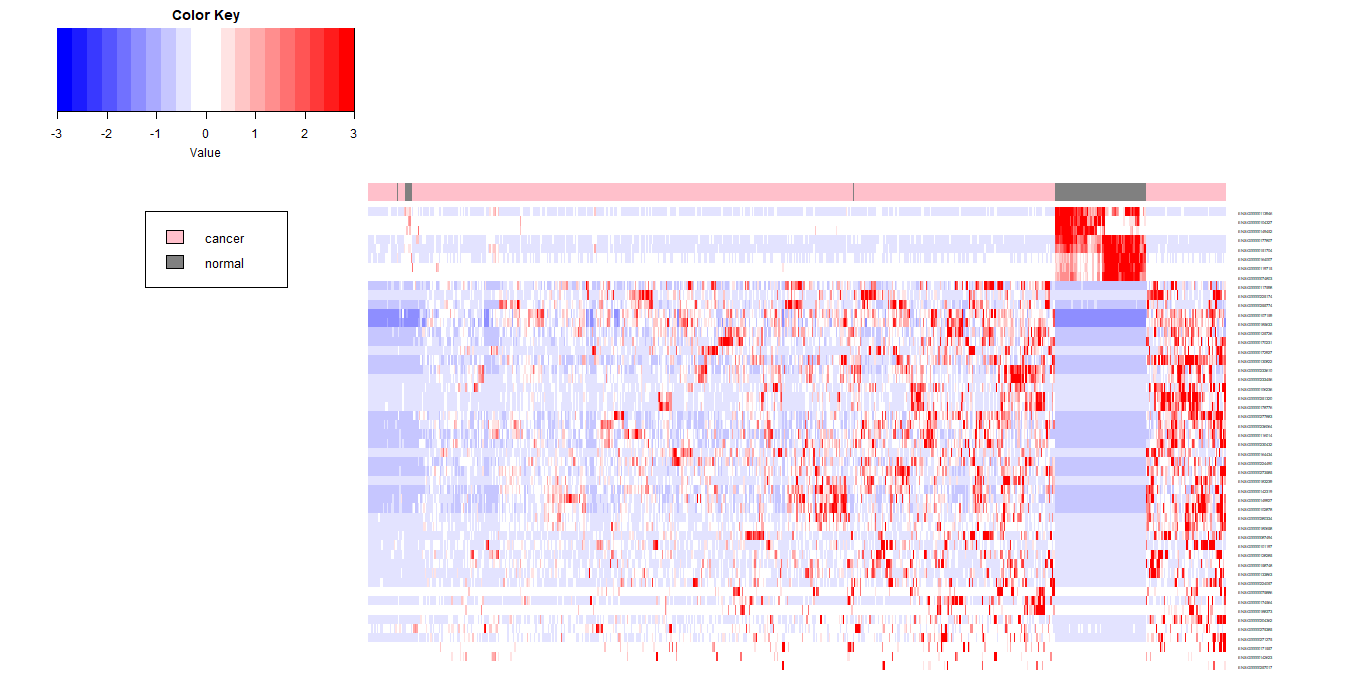
若限定在4以内,颜色进一步变淡
1 | heat_scaled_limit4 <- ifelse(heat_scaled>4,4,heat_scaled) |
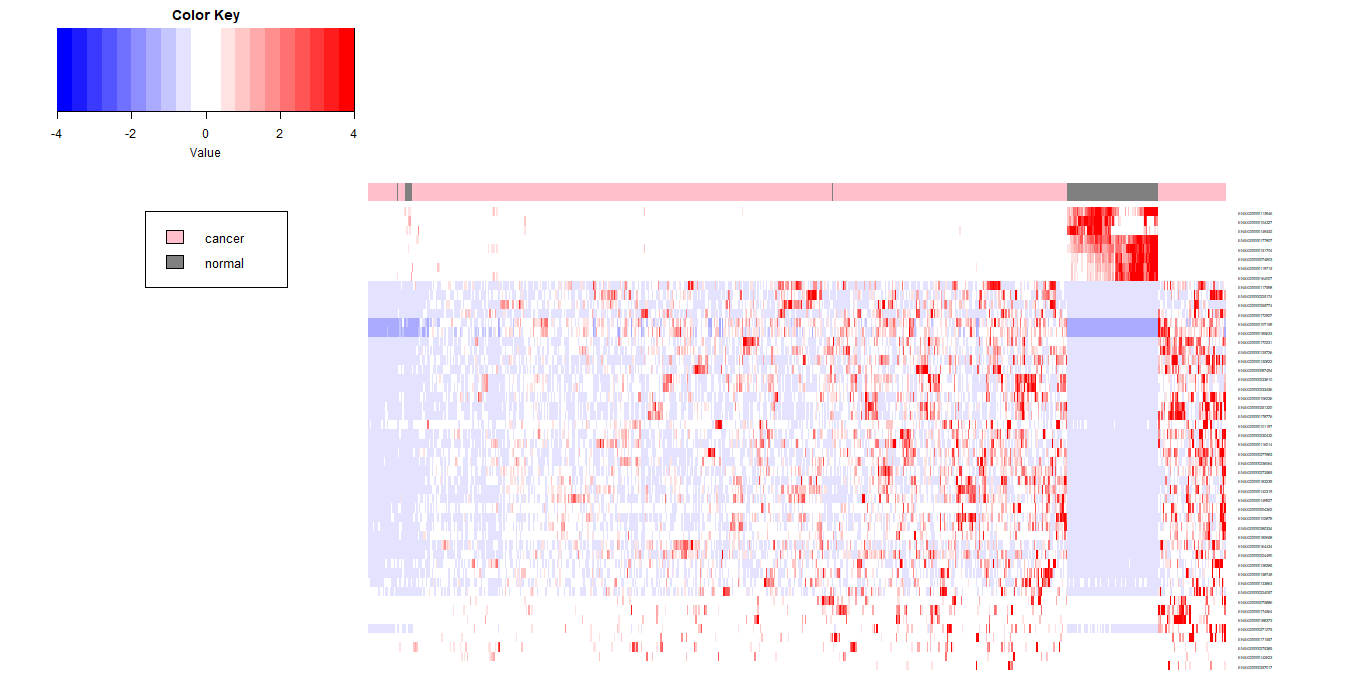
注意:对标准化后的值进行限定,基本不会对样本的聚类产生影响,但是会影响基因间的聚类,因此,产生的图像也会不同。
