GSEA(Gene Set Enrichment Analysis),是基因集富集分析,也是软件的名称,顾名思议,GSEA用于分析给定分组中差异表达的基因是否在给定的基因集中有富集。
下载GSEA软件:根据操作系统下载相应的版本并安装
软件主界面如下图:
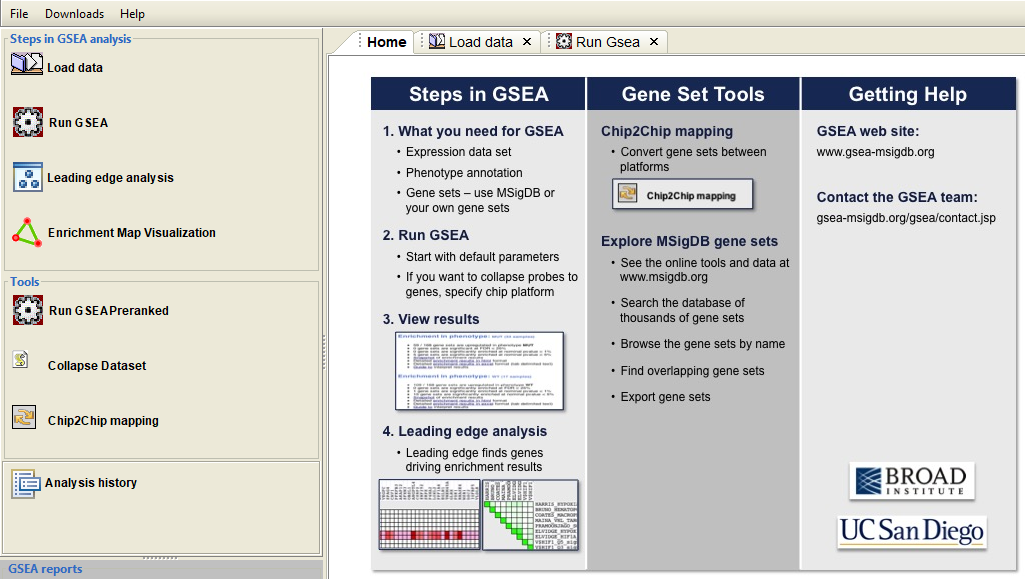
这个软件操作十分容易,简单明了,唯一的难度在于输入数据的准备,简单来说,有两个数据是必须提前准备好的,即表达数据与表型注释
表达数据格式:可以有多种输入格式,这里以TXT为例,我们先在R中将我们需要的数据提取并保存为CSV格式,处理完成后再保存为TXT即可。
1 | # 回顾一下前面的数据,差异表达筛选之后有4913个基因(result_select),注释后剩下4863个(result_select_annot) |
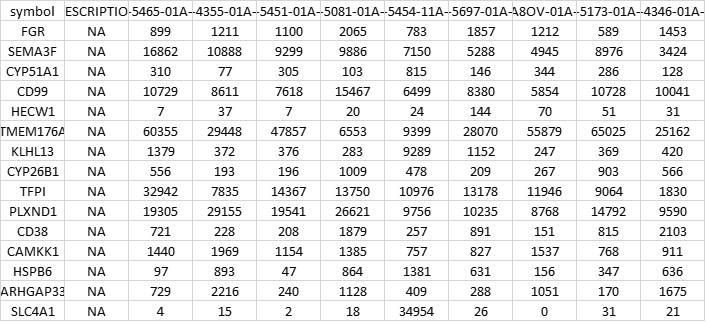
1 | # 下面准备表型文件 |
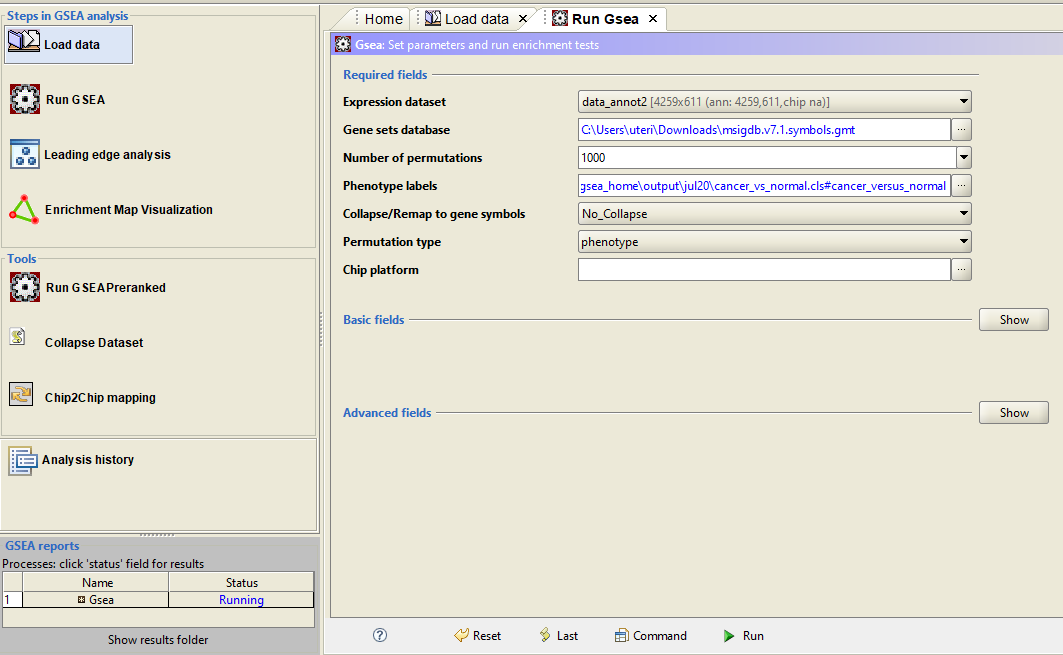
如上图,输入表达文件和表型文件,并且将下载好的基因集文件导入,就可以运行了,如果跑所有的基因集,需要耗费几分钟时间,运行完成后,生成一个网页,里面是对结果的描述。
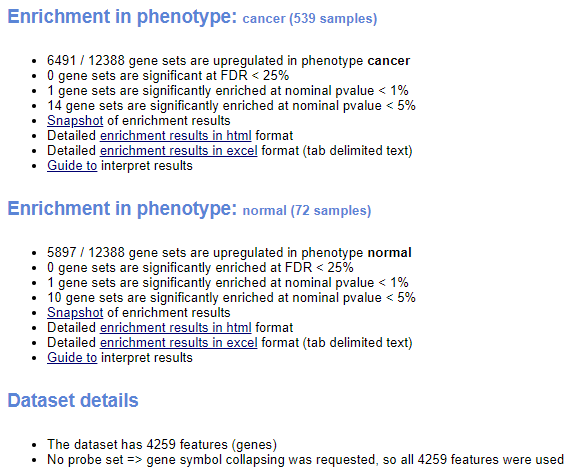
程序共运行了12388个基因集,使用了4259个基因。那么,富集结果怎么读呢?一般认为,|NES|>1,NOM p-val<0.05,FDR q-val<0.25的通路是显著富集的。
根据这个标准,富集结果并不理想,不论肿瘤或是正常组织中,都不存在符合以上标准的基因集。
当然,以上富集分析有一个硬伤,即只选取了差异表达的基因,并且由于注释的缘故,一部分基因被滤掉了,这可能导致富集结果出现偏差。我们也可以尝试使用所有的基因来做个分析。
1 | > dim(data_select) |
运行之后,结果是一致的,即没有显著富集的基因集。
