我们以TCGA-KIRC数据为例,来介绍肿瘤突变数据与临床数据的下载方法、预处理及可视化
一、数据下载与处理
在TCGA-GDC官网,在CASES选项下,选择CTGA-KIRC,这里共包含537个CASE,回到FILES选项下,选择Data Category[simple nucleotide variation]+Data Type[Masked Somatic Mutation]+Workflow Type[MuTect2 Variant Aggregation and Masking],至此,数据选择就完成了,点击Add Files to Cart,回到Cart,FILE=1,CASES=339,FILE SIZE=7.12 MB。下载Clinical/TSV文件,下载Download/Cart文件,前一个文件包含临床数据,后一个文件包含突变数据。
将这两个文件移至R的工作目录之下,使用命令对这两个tat.gz文件进行解压,
1 | tar -zxvf clinical.cart.2020-10-27.tar.gz # 解压后,得到三个文件:clinical.tsv,family_history.tsv,exposure.tsv |
在下一步操作之前,我们需要对下载好的数据作预处理,以使临床数据与突变数据能够通过Tumor_Sample_Barcode联系起来,分两步进行:
第一、打开clinical.tsv,将第二列“case_submitter_id”修改为“Tumor_Sample_Barcode”;
第二、打开mutect.maf,将“Tumor_Sample_Barcode”这一列中的内容修改为与clinical.tsv中的”Tumor_Sample_Barcode”一致,即只保留前12个字符,在EXCEL中可以新建一列,并使用[LEFT(text,12)]这个函数进行操作,操作完成后,使用CTRL+SHIFT+DOWN向下选择,再使用CTRL+D快速填充,填充完毕,将新列命名为”Tumor_Sample_Barcode”,并将旧的”Tumor_Sample_Barcode”删除,保存退出。
至此,数据下载与预处理就完成了,后面的过程会比较简单。
二、读取MAF文件并进行统计
2.1 MAF文件读取
下面的操作都将在RStudio/R中进行,代码如下:
1 | > setwd("D:/mutation/KIRC/mutect") # 切换至工作目录下 |
文件读取过程
1 | -Reading |
2.2 MAF文件统计
1 | > kirc # 查看MAF文件基本信息 |
1 | > getSampleSummary(kirc) # 对每一个样本的情况进行统计(336个样本) |
1 | > getGeneSummary(kirc) # 对每一个基因进行统计(9444个基因) |
1 | > getClinicalData(kirc) # 临床信息,即clinical.tsv文件中的内容,相同的case_id会输出一行,内容很长,不展示 |
1 | > getFields(kirc) # 显示MAF文件中的所有字段,第16列就是我们预先修改过的"Tumor_Sample_Barcode" |
1 | > write.mafSummary(maf=kirc, basename="kirc") # 输出一个kirc_sampleSummary.txt文件,内容就是getSampleSummary()输出的内容 |
三、突变的可视化
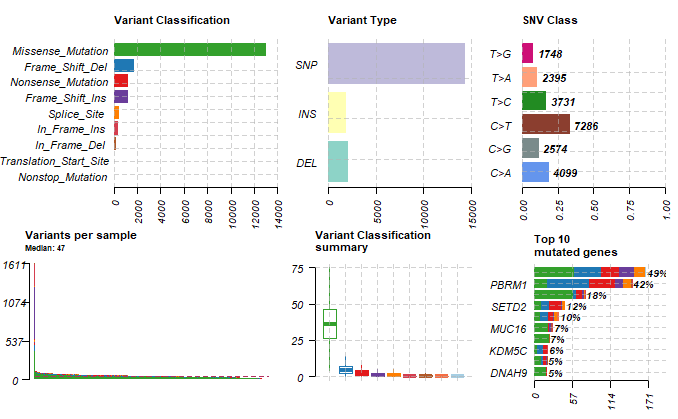
3.1 MAF文件汇总统计图
1 | > plotmafSummary(maf=kirc, rmOutlier=FALSE, addStat="median", dashboard=TRUE, titvRaw = FALSE) |
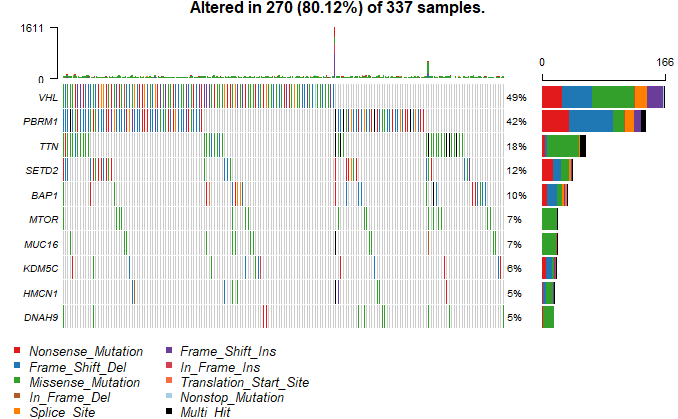
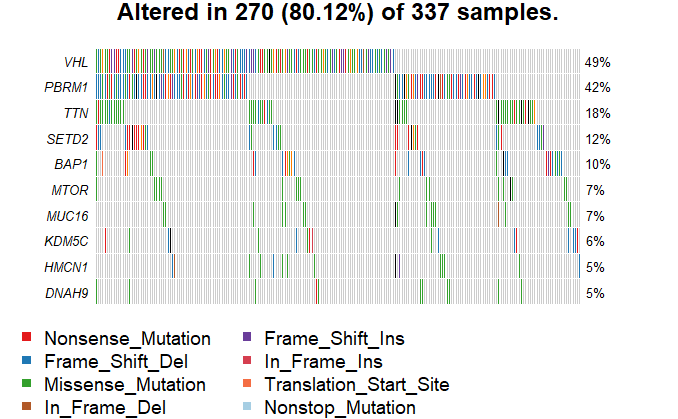
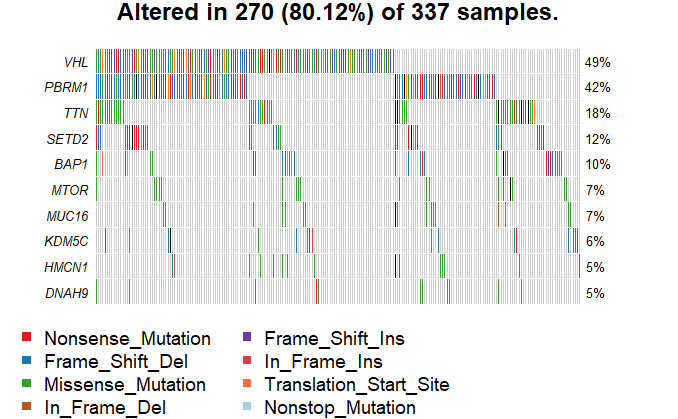
3.2 ONCOPLOT(瀑布图)
1 | oncoplot(maf=kirc,top=10,fontSize = 0.6,showTumorSampleBarcodes = F) |
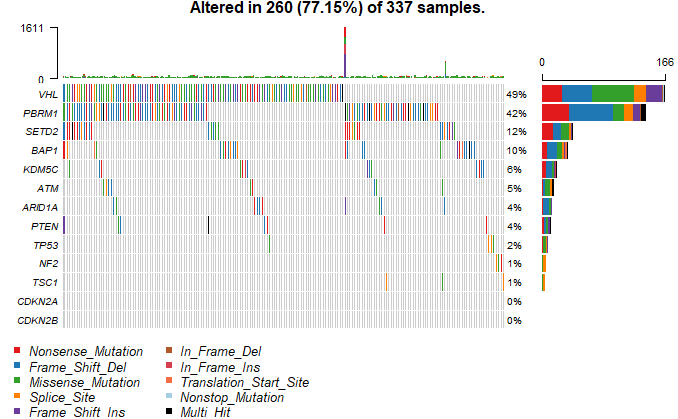
若对某些基因感兴趣,则使用genes参数指定基因,如下:
1 | oncoplot(maf=kirc,genes=c("VHL","PBRM1","SETD2","BAP1","KDM5C","TP53","ATM","ARID1A","PTEN","TSC1","NF2","CDKN2A","CDKN2B"),fontSize = 0.6,showTumorSampleBarcodes = F) |
3.3 基因突变共存与排斥
1 | > somaticInteractions(maf=kirc,top=10,pvalue = c(0.01,0.05),fontSize = 0.5) |
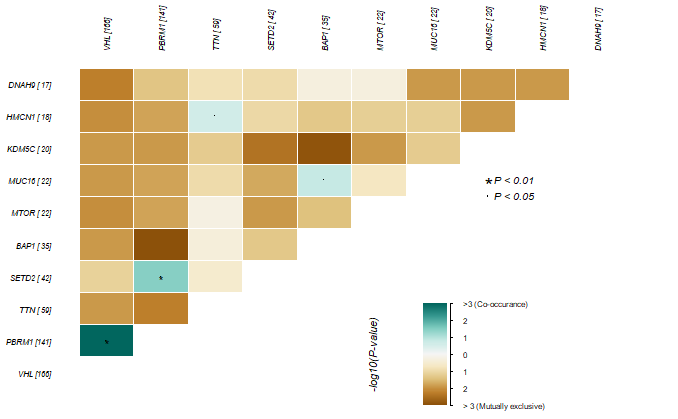
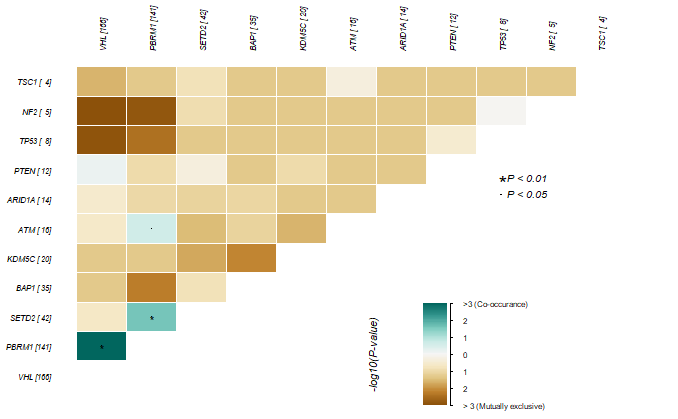
同理,也可以指定感兴趣的基因
1 | > somaticInteractions(maf=kirc,genes=c("VHL","PBRM1","SETD2","BAP1","KDM5C","TP53","ATM","ARID1A","PTEN","TSC1","NF2","CDKN2A","CDKN2B"),pvalue = c(0.01,0.05),fontSize = 0.5) |
3.4 文字云
1 | > geneCloud(input=kirc,top=10) |

3.5 ONCOSTRIP
1 | > oncostrip(maf=kirc,top=10) # oncostrip与oncoplot非常相似,但没有右侧的统计结果,按需选择吧 |
或按一组基因作图
1 | oncostrip(maf=kirc,genes=c("VHL","PBRM1","SETD2","BAP1","KDM5C","TP53","ATM","ARID1A","PTEN","TSC1","NF2","CDKN2A","CDKN2B")) |
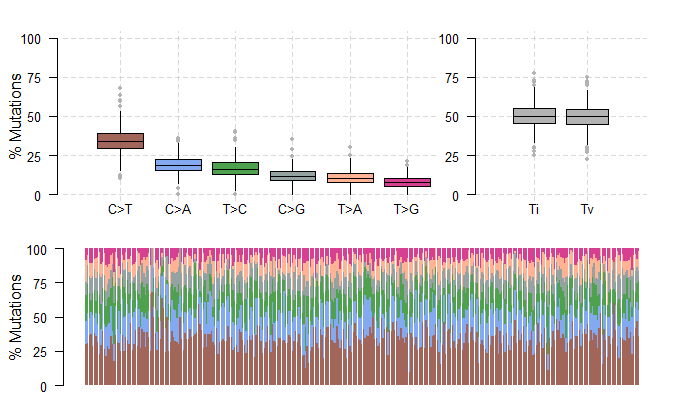
3.6 转换(transition)和颠换(transversion)
1 | > titv(maf=kirc, plot=TRUE, useSyn=TRUE) # titv函数的结果是对SNV_Class的另外一种诠释 |
3.7 LOLLIPOPPLOT
1 | > lollipopPlot(maf=kirc, gene="TP53", AACol="HGVSp_Short", showMutationRate=TRUE) |
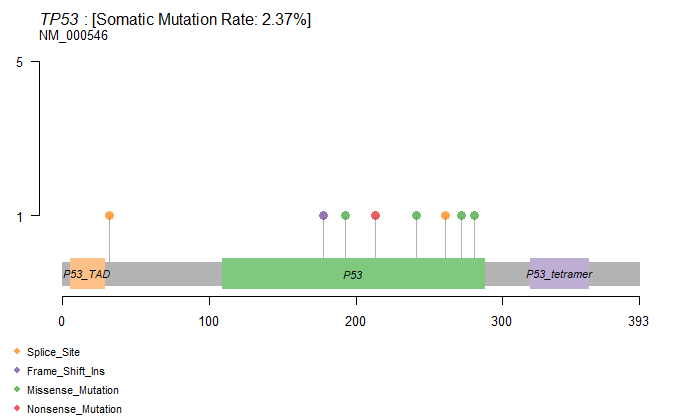
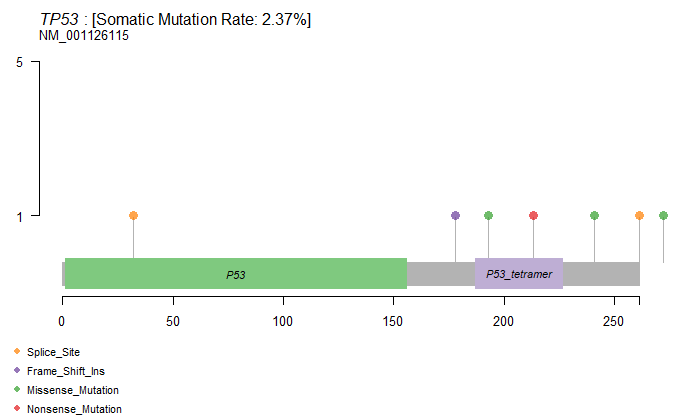
上面提示有报错信息,原因是TP53这个基因有8个已知的转录本,程序自动选择了最长的转录本,即NM_000546,我们可以通过refSeqID或proteinID手动指定转录本,如:
1 | lollipopPlot(maf=kirc, gene="TP53",refSeqID="NM_001126115", AACol="HGVSp_Short", showMutationRate=TRUE) |
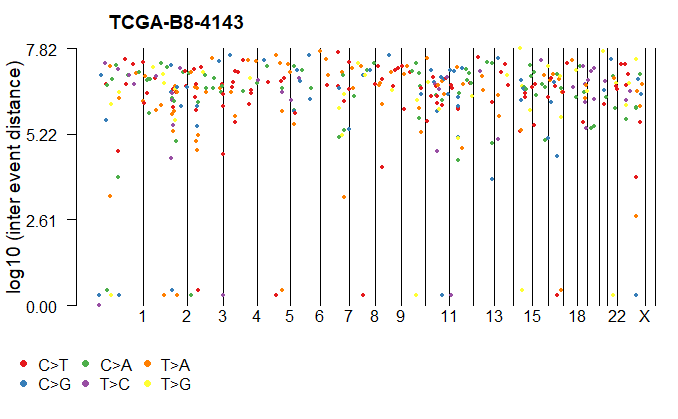
3.8 RAINFALLPLOT(降雨图)
1 | > rainfallPlot(maf=kirc, detectChangePoints=TRUE, pointSize=0.6) # 通过绘制染色体尺度的突变间距来展示hyper mutated genomic region |
3.9 TMB(肿瘤突变负荷)
1 | tcgaCompare(maf=kirc, cohortName="kirc") # cohortName是自定义的名称 |
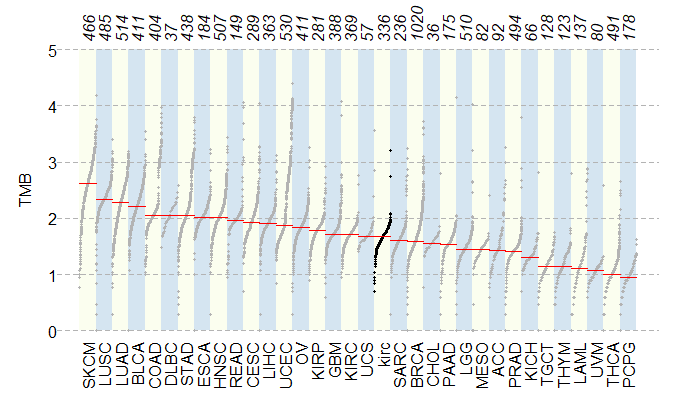
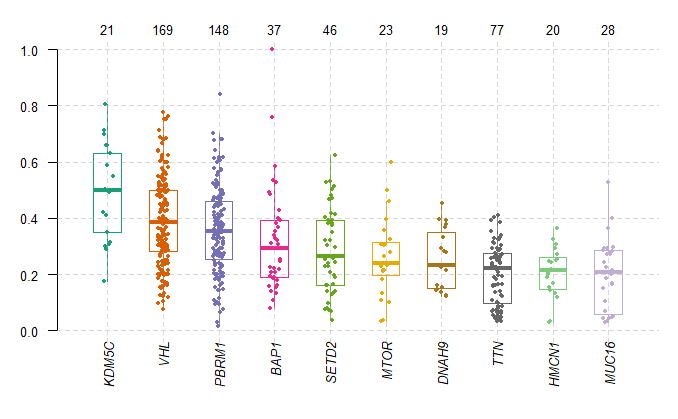
3.10 VAF(Variant Allele Frequency,变异等位基因频率)
1 | plotVaf(maf=kirc,top=10) # 默认top=10,可以手动指定,也可以使用genes指定基因名称 |
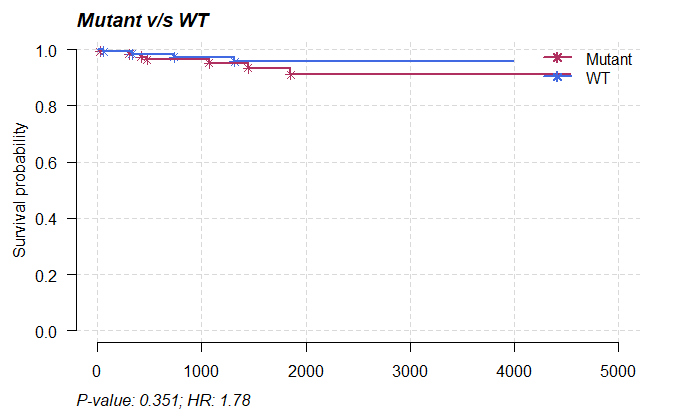
四、生存分析
生存分析是基于MAF文件中给定基因或样本的突变状态进行预测,可以通过参数genes指定一个或一组基因,可以通过参数samples指定样本,参数中的status需要特别注意,我这里使用的是”vital_status”,这个参数必须是“TRUE”/“FALSE”或者”1”/“0”,因此,需要在clinical.tsv中对相应的列进行修改,具体到不同的分析软件这一列的名称会大不相同。
1 | > mafSurvival(maf=kirc, genes=c("VHL"), time="days_to_last_follow_up", Status="vital_status", isTCGA=TRUE) |
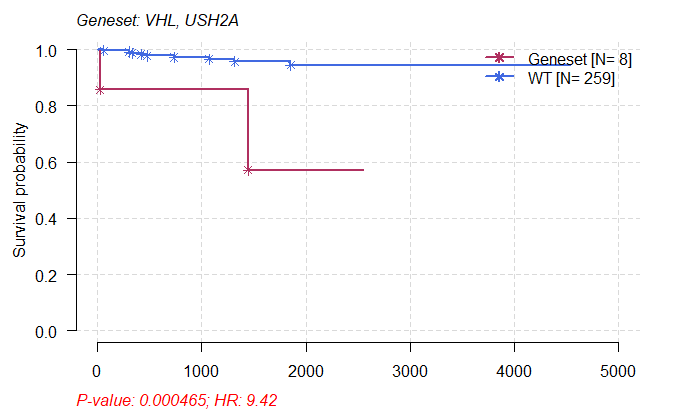
1 | > mafSurvGroup(maf=kirc,geneSet=c("SETD2","PBRM1"), time="days_to_last_follow_up", Status="vital_status") |
五、对比两个MAF文件
这一功能使得我们可以通过对临床数据进行分类而比较其差异,比如同一疾病中不同性别是否有显著差异,比如不同种族是否有显著差异,或者不同年龄阶段是否有显著差异,等等。下面,我们以性别将KIRC分为两类,
1 | kirc <- read.maf(maf="mutect.maf") |
至此,基于性别分类的两个MAF文件就准备好了,下面可以对这两组数据进行作图,
5.1 FORESTPLOT(森林图)
1 | comp <- mafCompare(m1=kirc.male, m2=kirc.female, m1Name="Male", m2Name="Female", minMut=5) |

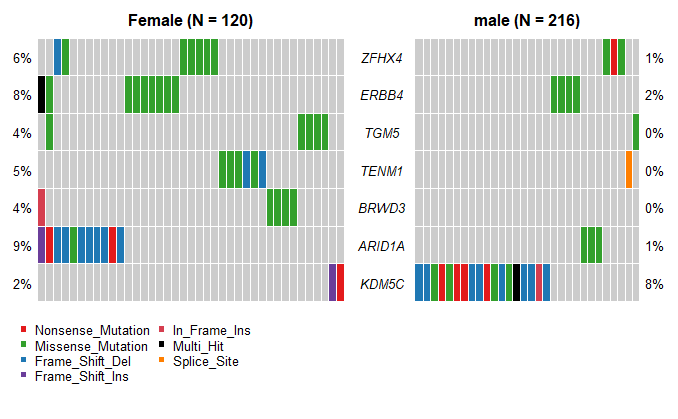
5.2 ONCOPLOT(双MAF瀑布图)
oncoplot中基因的选择,可根据mafCompare()的结果,选择有显著差异的基因,进行作图
1 | genes <- c("ZFHX4", "ERBB4", "TGM5", "TENM1", "BRWD3", "ARID1A", "KDM5C") |
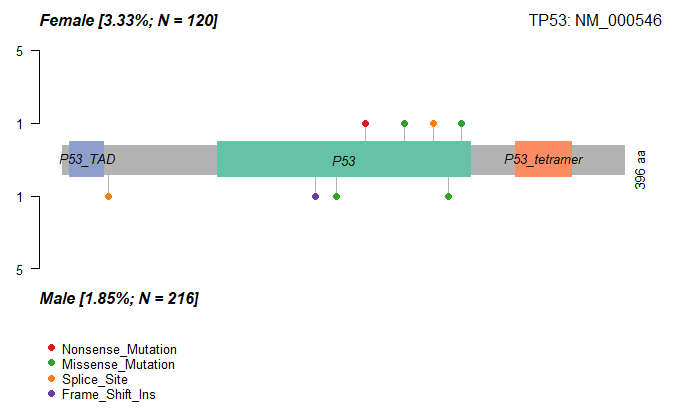
5.3 LOLLIPOPOLOT2(双MAF棒棒糖图)
1 | lollipopPlot2(m1=luad.female, m2=luad.male, m1_name="Female", m2_name="Male", gene="IQGAP2", AACol1 = "HGVSp_Short", AACol2 = "HGVSp_Short") |
六、信号通路富集
1 | > OncogenicPathways(maf = laml) |
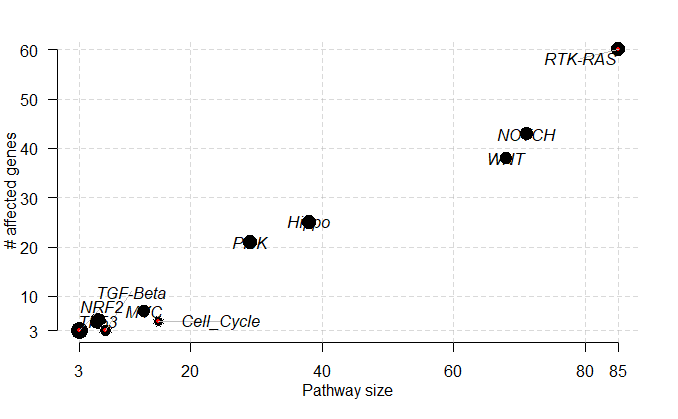
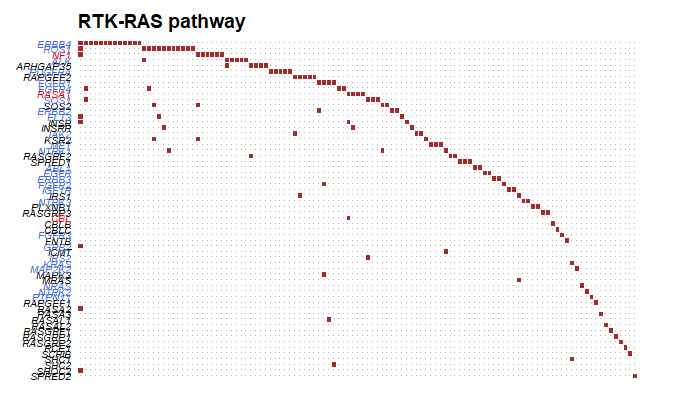
从上面输出的统计结果,可以选择感兴趣的信号通路进行作图,注意,图中的红色表示抑癌基因,蓝色表示致癌基因。
1 | > PlotOncogenicPathways(maf=kirc, pathways = "RTK-RAS") |
七、临床富集分析
clinicalEnrichment可以对指定的临床特征进行富集分析,比如指定”ajcc_pathologic_stage”,命令如下:
1 | > stage = clinicalEnrichment(maf=kirc, clinicalFeature = 'ajcc_pathologic_stage') |

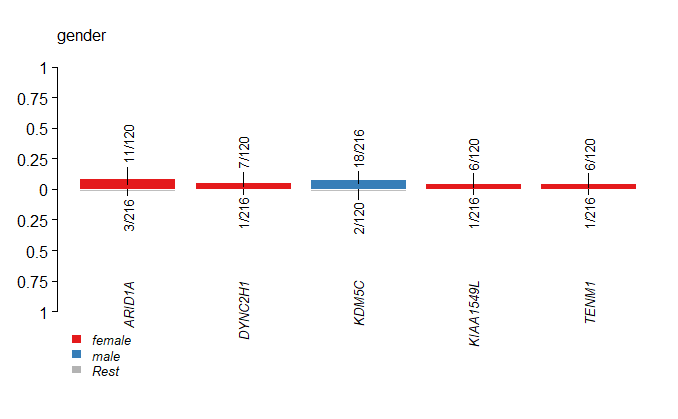
同理,也可对其他临床特征进行富集分析,比如性别
1 | > gender = clinicalEnrichment(maf=kirc, clinicalFeature = 'gender') |
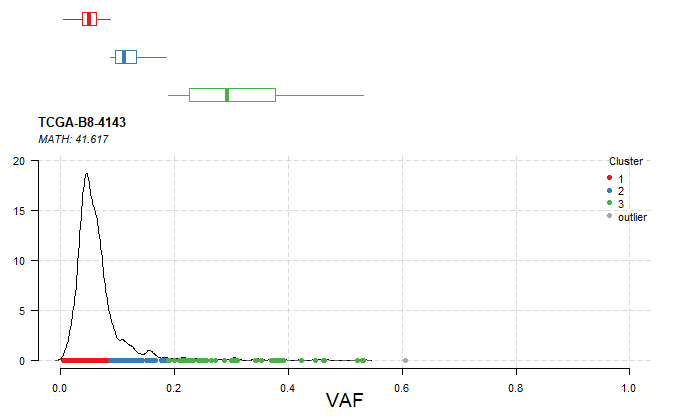
八、肿瘤异质性分析
一般而言,肿瘤是异质的,包含不同的亚型的细胞,可以通过inferHeterogeneity来对VAF进行评估以推断肿瘤异质性
1 | > inferHeterogeneity(maf=kirc) |
上面,对5个barcode进行了整体分析,下面,可以指定某一个Barcode来具体看一下,比如指定”TCGA-B8-4143”
1 | > barcode1 <- inferHeterogeneity(maf=kirc, tsb="TCGA-B8-4143") |