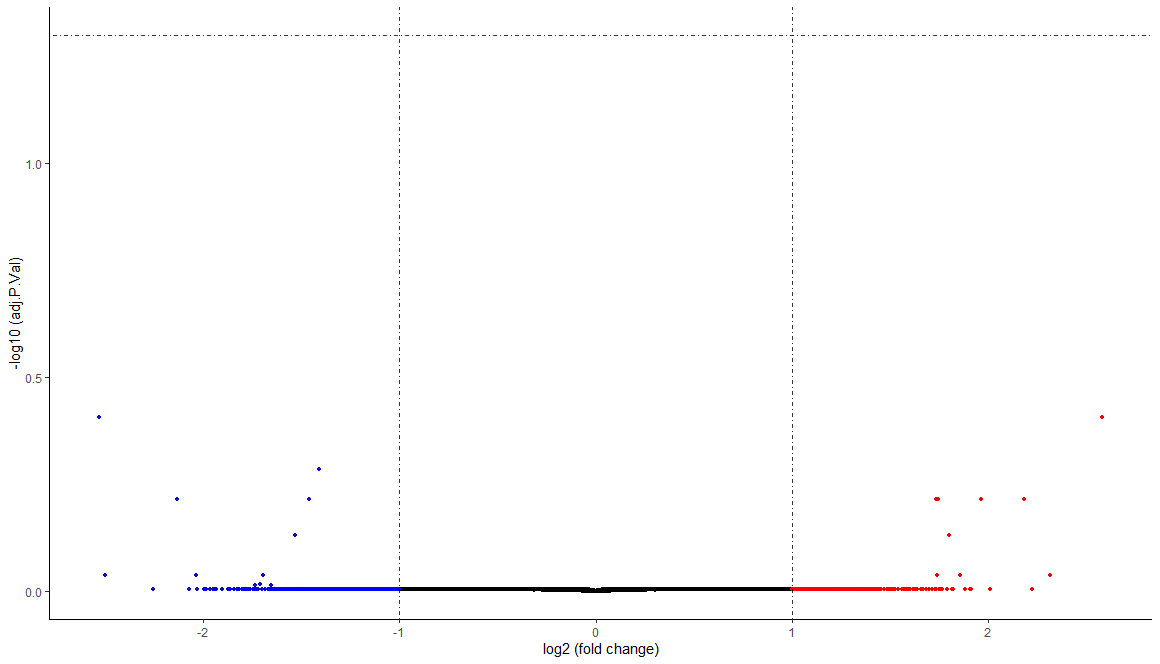
GEO学习笔记7 发表于 2020-08-01 | 分类于 生物信息 | | 热度 °C 分析GSE72922,23个样本,非配对,平台是GPL14951,然而,没有差异基因 123456789101112setwd("D:/GEO")library(GEOquery)gset <- getGEO('GSE72922',destdir=".",getGPL=F)gset <- gset[[1]]pdata <- pData(gset)dim(pdata)group_list <- c(rep('tumor',7),rep('normal',4),rep('tumor',2),rep('normal',7),rep('tumor',3))group_list <-factor(group_list)group_list <- relevel(group_list, ref='tumor')group_listexprSet <- exprs(gset)boxplot(exprSet,outline=FALSE,notch=T,col=group_list,las=2) 123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129# NORMALIZATION ----# NO NEED NORMALIZATIONlibrary(limma) #exprSet <- normalizeBetweenArrays(exprSet)#boxplot(exprSet,outline=FALSE,notch=T,col=group_list,las=2)# LOG2 TRANSFORM ----ex <- exprSetqx <- as.numeric(quantile(ex, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))LogC <- (qx[5] > 100) || (qx[6]-qx[1] > 50 && qx[2] > 0) || (qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)if (LogC) { ex[which(ex <= 0)] <- NaNexprSet <- log2(ex) }dim(exprSet)# ANNOTATION ----gpl <- getGEO('GPL14951',destdir = './')gpl <- getGEO(filename = './GPL14951.soft')GPL14951 <- GEOquery::Table(gpl)#probeset <- rownames(exprSet)#probe2symbol <- data.frame(probeset,symbol=GPL14951$Symbol,stringsAsFactors = F)library(dplyr)x1 <- data.frame(probeset=rownames(exprSet))x2 <- data.frame(probeset=GPL14951$ID,symbol=GPL14951$Symbol)probe2symbol <- x1 %>% inner_join(x2,by='probeset')dim(probe2symbol)head(probe2symbol)# REMOVE NA ----library(dplyr)library(tibble)exprSet <- as.data.frame(exprSet)exprSet <- exprSet %>% rownames_to_column(var='probeset') %>% inner_join(probe2symbol,by='probeset') %>% select(-probeset) %>% select(symbol,everything()) %>% mutate(rowMean =rowMeans(.[grep('GSM', names(.))])) %>% filter(symbol != 'NA') %>% arrange(desc(rowMean)) %>% distinct(symbol,.keep_all = T) %>% select(-rowMean) %>% column_to_rownames(var = 'symbol')dim(exprSet)exprSet <- na.omit(exprSet)dim(exprSet)# DIFF EXPRESS ----# NO PAIRINGdesign <- model.matrix(~ group_list)fit <- lmFit(exprSet,design)fit <- eBayes(fit) allDiff_pair <- topTable(fit,adjust='BH',coef='group_listnormal',number=Inf,p.value=0.05)> dim(allDiff_pair)[1] 0 0 # 没有差异基因,不必往下分析# PLOT ----data_plot <- as.data.frame(t(exprSet))data_plot <- data.frame(pairinfo=pairinfo, group=group_list, data_plot,stringsAsFactors = F)library(ggplot2)ggplot(data_plot, aes(group,EEF1A1,fill=group)) + geom_boxplot() + geom_point(size=2, alpha=0.5) + geom_line(aes(group=pairinfo), colour='black', linetype='11') + xlab('') + ylab(paste('Expression of ','EEF1A1'))+ theme_classic()+ theme(legend.position = 'none')# COMBINE PLOT ----library(dplyr)library(tibble)allDiff_arrange <- allDiff_pair %>% rownames_to_column(var='genesymbol') %>% arrange(desc(abs(logFC)))genes <- allDiff_arrange$genesymbol[1:10]genesplotlist <- lapply(genes, function(x){ data =data.frame(data_plot[,c('pairinfo','group')],gene=data_plot[,x]) ggplot(data, aes(group,gene,fill=group)) + geom_boxplot() + geom_point(size=2, alpha=0.5) + geom_line(aes(group=pairinfo), colour='black', linetype='11') + xlab('') + ylab(paste('Expression of ',x))+ theme_classic()+ theme(legend.position = 'none')})library(cowplot)plot_grid(plotlist=plotlist, ncol=5,labels = LETTERS[1:10])# HEATMAP ----library(pheatmap)allDiff_pair2 <- topTable(fit,adjust='BH',coef='group_listnormal',number=Inf,p.value=0.05,lfc = 1) dim(allDiff_pair2)heatdata <- exprSet[rownames(allDiff_pair2),]heat <- na.omit(heatdata)annotation_col <- data.frame(group_list)rownames(annotation_col) <- colnames(heatdata)pheatmap(heat, cluster_rows = TRUE, cluster_cols = TRUE, annotation_col =annotation_col, annotation_legend=TRUE, show_rownames = F, show_colnames = T, scale = 'row', color =colorRampPalette(c('blue', 'white','red'))(100))# VOLCANO PLOT ----library(ggplot2)library(ggrepel)library(dplyr)data <- topTable(fit,adjust='BH',coef='group_listnormal',number=Inf) dim(data)data$significant <- as.factor(data$adj.P.Val<0.05 & abs(data$logFC) > 1)data$gene <- rownames(data) ggplot(data=data, aes(x=logFC, y =-log10(adj.P.Val),color=significant)) + geom_point(alpha=0.8, size=1.2,col='black')+ geom_point(data=subset(data, logFC > 1),alpha=0.8, size=1.2,col='red')+ geom_point(data=subset(data, logFC < -1),alpha=0.6, size=1.2,col='blue')+ labs(x='log2 (fold change)',y='-log10 (adj.P.Val)')+ theme(plot.title = element_text(hjust = 0.4))+ geom_hline(yintercept = -log10(0.05),lty=4,lwd=0.6,alpha=0.8)+ geom_vline(xintercept = c(1,-1),lty=4,lwd=0.6,alpha=0.8)+ theme_bw()+ theme(panel.border = element_blank(), panel.grid.major = element_blank(), panel.grid.minor = element_blank(), axis.line = element_line(colour = 'black')) 12345678910111213141516# ENRICH library(clusterProfiler)gene <- rownames(allDiff_pair)gene <- bitr(gene, fromType='SYMBOL', toType='ENTREZID', OrgDb='org.Hs.eg.db')de <- gene$ENTREZIDgo <- enrichGO(gene = de, OrgDb = 'org.Hs.eg.db', ont='all')library(ggplot2)dotplot(go, split='ONTOLOGY') +facet_grid(ONTOLOGY~., scale='free')EGG <- enrichKEGG(gene= gene$ENTREZID, organism = 'hsa', pvalueCutoff = 0.05)dotplot(EGG)test <- data.frame(EGG)head(test)[,1:7]browseKEGG(EGG, 'hsa04151') 本文作者:括囊无誉 本文链接: GEO/GEO07/ 版权声明: 本博客所有文章均为原创作品,转载请注明出处! ------ 本文结束 ------ 坚持原创文章分享,您的支持将鼓励我继续创作! Donate WeChat Pay