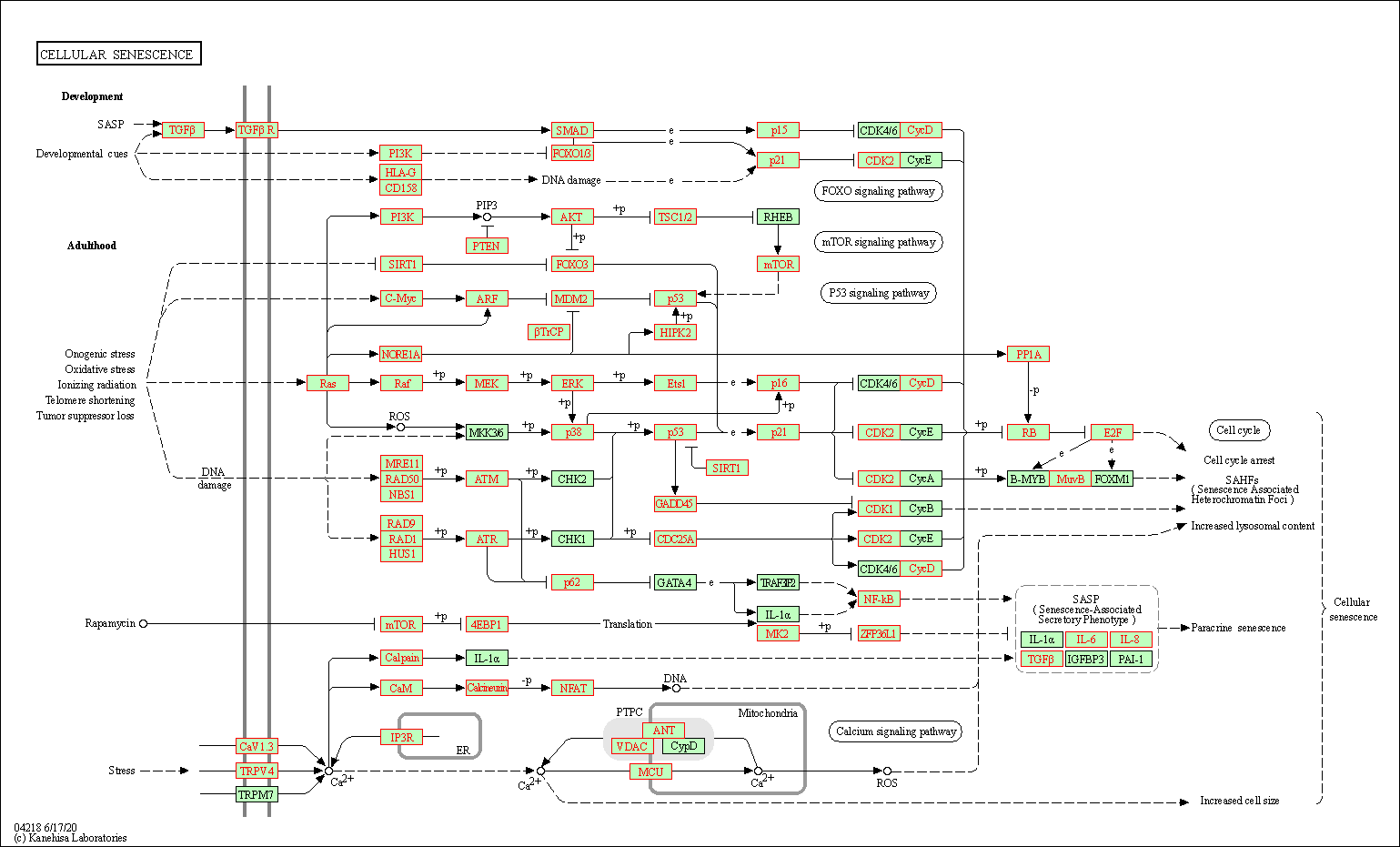
本文是学习过程,参考教程是《来完成你的生信作业,这是最有诚意的GEO数据库教程》,本文分析GSE32575,共36个配对样本,18个处理前,18个处理后。
1 | > setwd("D:/GEO") |
1 | library(limma) # 使用limma进行Normalization |
1 | # 对数据进行LOG转换,否则数据太过分散 |
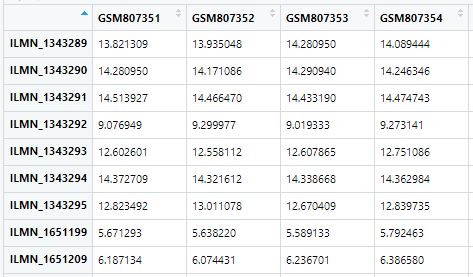
转换之后,行名是探针名称,需要先转换为基因名称
1 | > index <- gset@annotation |
其实,注释的过程中有些基因是没有名称的,会被标记为NA,另外也可能有重复的,即多个探针可能对应同一基因,因此,需要去掉重复与NA。
1 | > exprSet <- exprSet %>% |
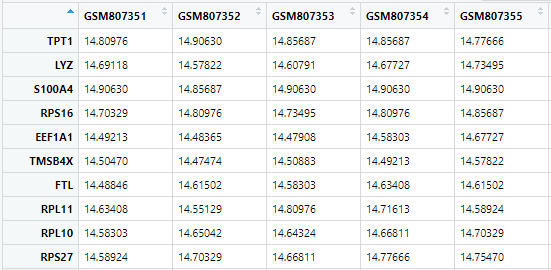
到此,数据就准备好了,下面进行差异分析
1 | > pairinfo <- factor(rep(1:18,2)) |
作图验证一下,作图之前需要对数据格式进行调整,以适应ggplot2对输入数据的要求,即行是样品,列是基因
1 | data_plot <- as.data.frame(t(exprSet)) |
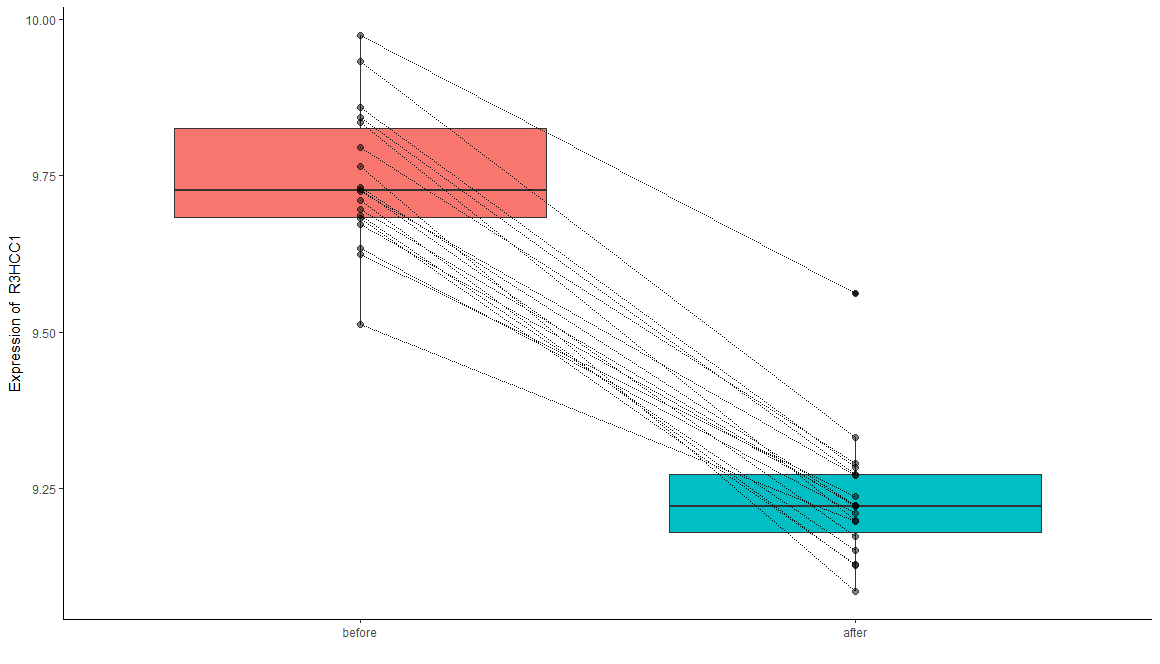
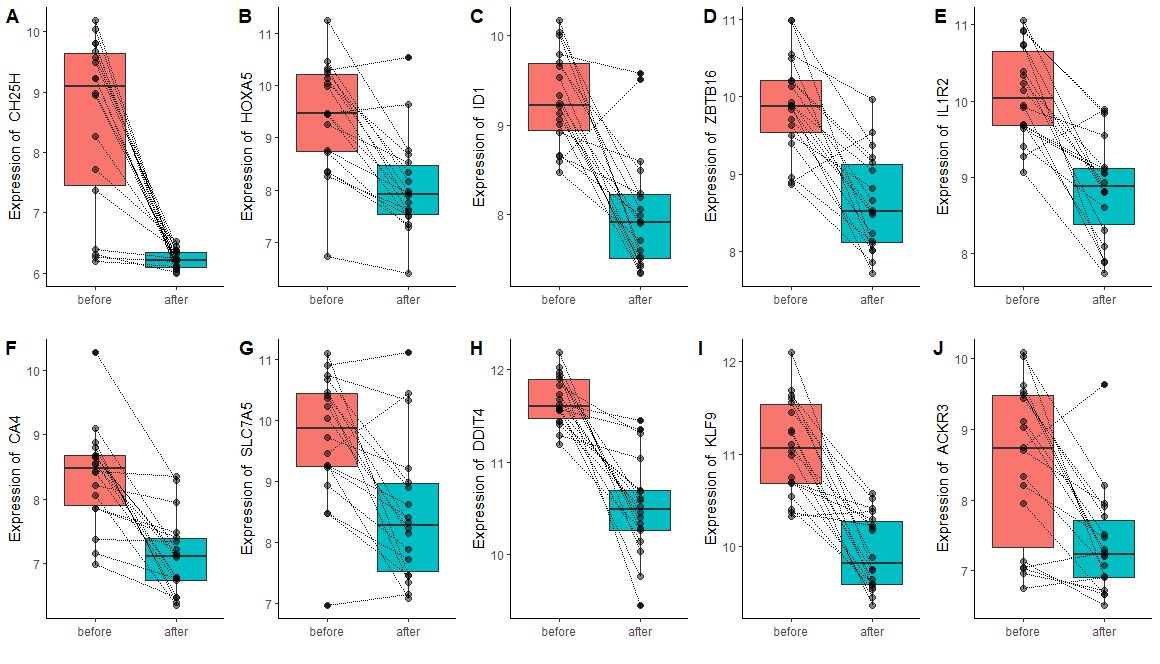
批量作图,挑差异最大的10个基因
1 | > library(dplyr) |
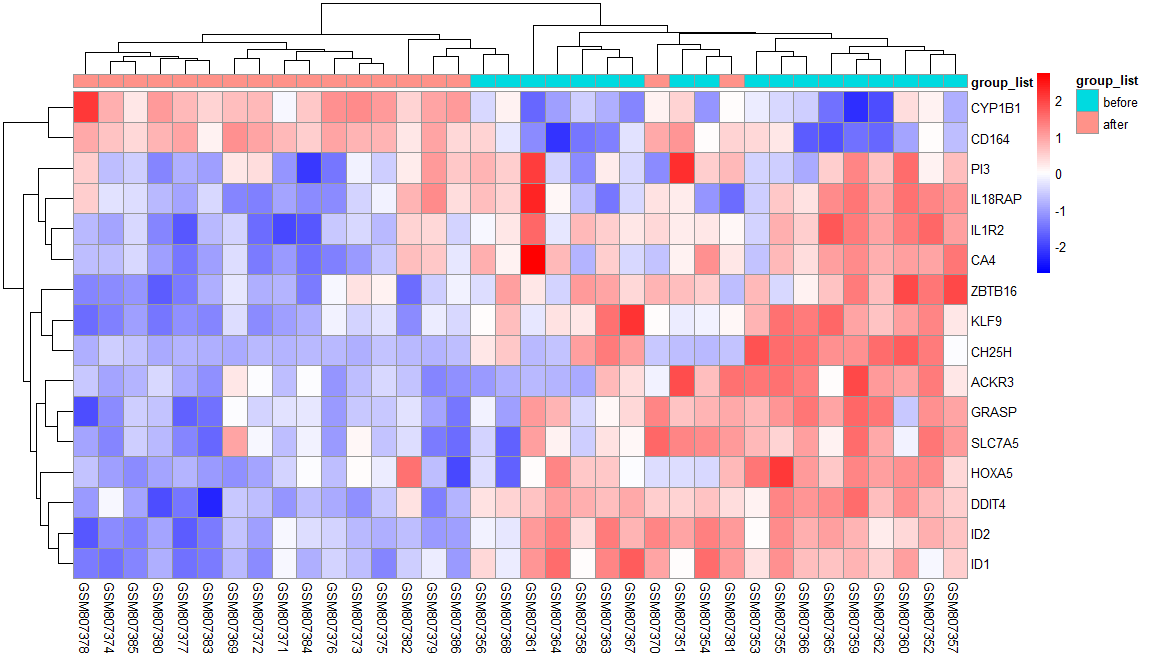
画热图
1 | library(pheatmap) |
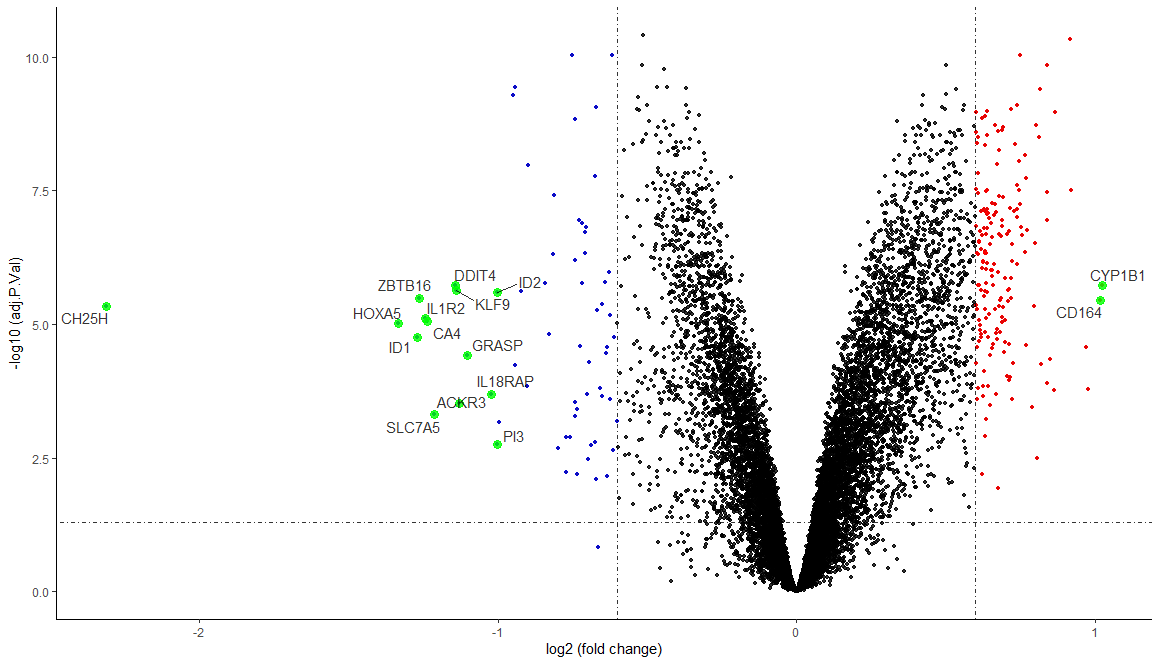
绘制火山图
1 | > library(ggplot2) |
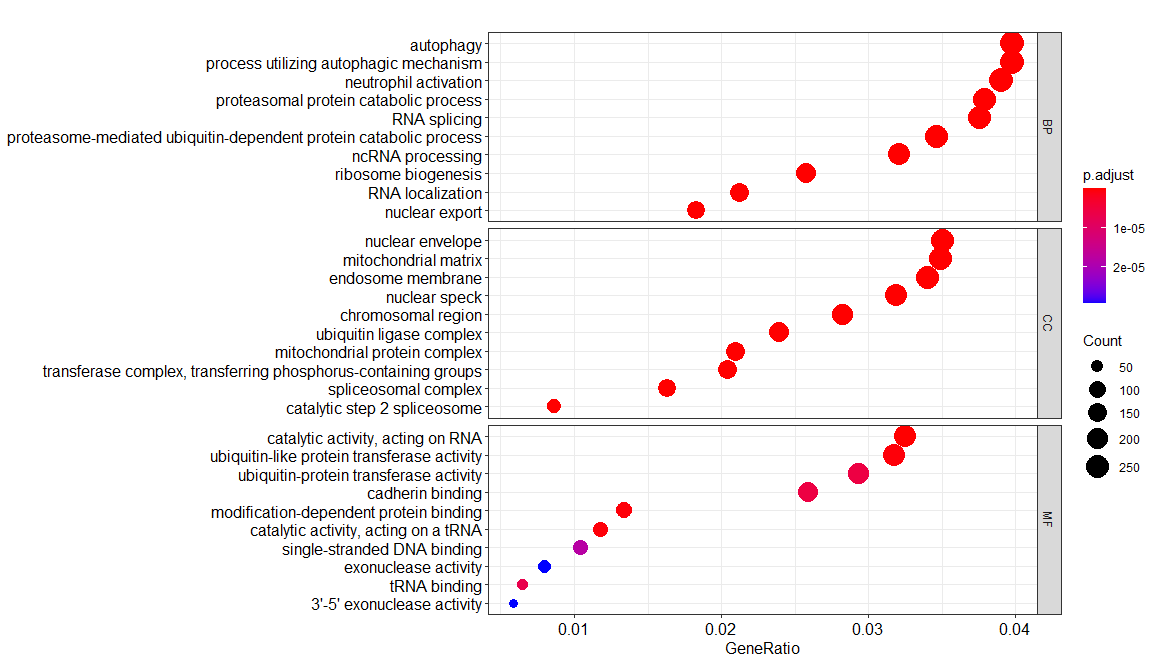
富集分析
1 | > library(clusterProfiler) |
1 | > EGG <- enrichKEGG(gene= gene$ENTREZID, |
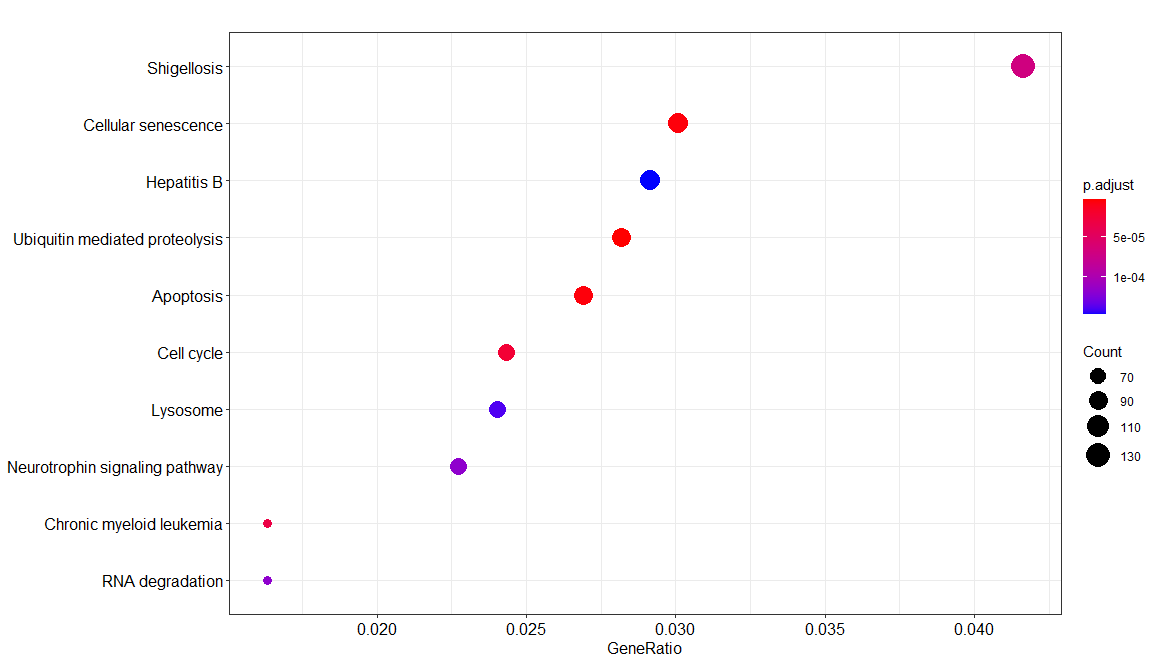
1 | > test <- data.frame(EGG) |